
Abstract
Persistent eosinophil activation in both the upper and lower airway mucosa is a central feature of aspirin-exacerbated respiratory disease (AERD). Eosinophil activation and survival are profoundly influenced by interleukin 5 (IL-5) and its receptor, IL-5R. In patients susceptible to allergic disorders, IL-5 receptor α (IL5RA) polymorphisms have been reported; however, an association with AERD remains unclear. We hypothesize that IL5RA polymorphisms may contribute to eosinophil activation in AERD patients. We recruited 139 AERD patients, 171 aspirin-tolerant asthma patients and 160 normal controls. IL5RA polymorphisms (−5993G>A, −5567C>G and −5091G>A) were genotyped and functional activity of polymorphism was assessed by luciferase reporter assay and electrophoretic mobility shift assay (EMSA). There was no significant difference in the genotype frequency of the three polymorphisms among the three groups. AERD patients carrying the AA genotype at −5993G>A had a significantly higher presence of serum-specific immunoglobulin E (IgE) to staphylococcal enterotoxin A (P=0.008) than those with the GG/GA genotype. In vitro, the −5993A allele had a higher promoter activity compared with the −5993G allele in human mast cell (HMC-1; P=0.030) and human promyelocytic leukemia (HL-60; P=0.013) cells. In EMSA, a −5993A probe produced a specific shifted band than the −5993G had. These findings suggest that a functional polymorphism in IL5RA may contribute to eosinophil and mast cell activation along with specific IgE responses to staphylococcal enterotoxin A in AERD patients.
Similar content being viewed by others

Introduction
Aspirin-exacerbated respiratory disease (AERD) refers to the development of bronchoconstriction in individuals with chronic asthma after exposure to aspirin and most other nonsteroidal anti-inflammatory drugs. Patients with AERD experience progressively worsening nasal and respiratory symptoms that are mostly associated with severe markers than aspirin-tolerant asthma (ATA) patients.1, 2 Nasal polyp and sinus pathology of AERD patients shows intense infiltration of eosinophils and evidence of eosinophilic activation, through the release of eosinophil granule proteins and eotaxin production.3, 4, 5 Similarly, following aspirin challenge, bronchoalveolar lavage fluid from AERD patients contained increased numbers of eosinophils and eosinophil cationic protein (ECP) compared with other types of asthma patients.6 The infiltration of activated eosinophils in the respiratory mucosa aggravates airway inflammation by eosinophil recruitment as well as airway remodeling in AERD patients.7, 8
The production and function of eosinophils is markedly influenced by interleukin-5 (IL-5) via the IL-5 receptor (IL-5R). The IL-5R, a heterodimer consisting of α-subunit (IL5RA on 3p26-p24) and β-subunit, is expressed on eosinophils, basophils and mast cells.9, 10 Single-nucleotide polymorphisms (SNPs) of the IL5RA gene have been documented in asthma and other allergies, and are thought to cause enhanced levels of peripheral blood eosinophils and ECP.11, 12, 13 Increased numbers of IL5RA mRNA-positive cells in the bronchial biopsies of asthmatic subjects have been reported.14 Recent attention has focused on the development of a humanized monoclonal antibody targeting the IL5RA as a therapeutic agent to treat eosinophil-dominant asthma. This works by reducing the level of peripheral blood eosinophils and ECPs in mild asthmatics,15 thus demonstrating the critical role of IL5RA in the development of IL-5-mediated eosinophil activation in the inflammation of the upper and lower airways.
Numerous studies have emphasized the multifactorial nature of AERD.16 Interactions of genetic and environmental factors are pivotal in the pathophysiology of AERD. Staphylococcal superantigens are well-known environmental factors modulating the severity of asthma, through decreased lung function and eosinophilic inflammation.4, 17, 18 Particularly, serum levels of specific immunoglobulin E (IgE) to staphylococcal superantigens were higher in AERD patients, and those with detectable levels of specific IgE to these superantigens had increased airway hyperresponsiveness.19
In this study, we hypothesized that IL5RA gene polymorphisms may be involved in eosinophil activation in AERD patients. We designed a case–control study to identify potential interactions between IL5RA gene polymorphisms and the AERD phenotype in a Korean population. The functional effects of the IL5RA polymorphism were assessed by a luciferase reporter assay and an electrophoretic mobility shift assay (EMSA).
Materials and methods
Study subjects
A total of 139 adult patients with AERD were recruited at the Ajou University Hospital, Suwon, South Korea. The diagnosis of AERD was based on a positive response to a lysine aspirin bronchoprovocation test, which was performed on all the asthmatic subjects according to previously described methods.20 A change of forced expiratory volume after 1 s (FEV1) was noted up to 7 h after the last lysine aspirin challenge. In all, 171 patients with ATA were recruited from those who were identified as negative responders to the lysine aspirin bronchoprovocation test. The 160 normal controls (NC) included in this group had no history of respiratory symptoms and showed normal results on simple chest radiograms and spirometry tests. Skin prick testing was performed for 12 common aero-allergens (Bencard, West Sussex, UK). Atopy was defined as one or more positive reactions to common inhalant allergens. The presence of chronic rhinosinusitis and nasal polyps was evaluated by paranasal sinus X-ray and rhinoscopy. Serum total IgE, ECP and specific IgE to staphylococcal enterotoxins A and B and toxic shock syndrome toxin 1 were measured using the UniCAP system (Phadia, Valinge, Sweden) as per the described methodology.21 We defined a specific IgE level of >0.35 KU l−1 as positive, according to the manufacturer’s guidelines. All subjects provided informed consent, and the institutional review board of Ajou University Hospital approved the study.
SNP genotyping
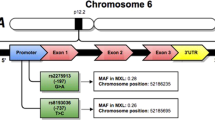
In this study, we selected −5993G>A (rs7622183), −5567C>G (rs3806681) and −5091G>A (rs2290608) SNPs for genotyping with AERD, ATA and NC. SNP genotyping was performed using a primer extension method and the SNAPshot ddNTP primer extension kit (Applied Biosystems, Foster City, CA, USA). Sequences of the amplification and extension primers were as follows: for −5993G>A, forward primer 5′-ACGCTGACTGTACTTGCAC-3′, reverse primer 5′-GATTCCTTCTGGGGAATTT-3′ and extension primer 5′-TGCTCTGTCTGAGACTCTCCCAGTC-3′; for the −5567C>G polymorphism, forward primer 5′-AGGAGGTTTGCTGAAATTG-3′, reverse primer 5′-GGCAACACTGACACATGAT-3′ and extension primer 5′-ACACATATTTACTTGAGGGTAC-3′, and for the −5091G>A polymorphism, forward primer 5′-TTCTTTCGTTTGACAGCAG-3′, reverse primer 5′-TCTGTGGTGAGTGAAGCAT-3′ and extension primer 5′-ATCATGAAGGAAGCTGCCTGAGA-3′.
Cell culture and preparation of the IL5RA constructs
Human mast cell (HMC-1) and human promyelocytic leukemia (HL-60) cell lines were cultured in Iscove’s modified Dulbecco’s medium (Gibco, Grand Island, NY, USA), supplemented with 10% heat-inactivated fetal bovine serum, 100 U ml−1 penicillin G sodium and 100 μg ml−1 of streptomycin sulfate (Gibco), at 37 °C in a 5% CO2 incubator.
A 1143-bp fragment of the human IL5RA gene was amplified from the genomic DNA of −5993GG homozygous subjects by PCR using the following primers: forward primer 5′-CACACGCTGACTGTACTTGCAC-3′ and reverse primer 5′-GGGCATTGAGAACGAACCTTA-3′. PCR products were gel-purified with an agarose gel purification kit (iNtRON, Deajeon, Korea) and ligated into the TA vector (Invitrogen, Carlsbad, CA, USA). Next, the plasmid was digested with SacI and XhoI restriction enzymes (Takara, Shuzo, Japan) and ligated into a similarly digested reporter gene pGL3-basic vector (Promega, Madison, WI, USA). The −5993A construct was generated by site-directed mutagenesis using the −5993G construct as a template. Site-directed mutagenesis was performed using the overlap-extension PCR mutagenesis technique.22 Before transfection, the constructs were verified by direct sequencing.
Transient transfection and dual luciferase assay
Construct plasmids were transfected into HMC-1 and HL-60 cells using an MP-100 microporator (Digital Biotechnology, Seoul, Korea) and MPK-1096 (Digital Biotechnology) according to the manufacturer’s instructions. Twelve-well plates were filled with 1 ml Iscove’s modified Dulbecco’s medium in culture, containing 10% fetal bovine serum. Plates were preincubated in a humidified incubator at 37 °C with 5% CO2. HMC-1 (2 × 105 cells per well) and HL-60 (5 × 105 cells per well) cells, reporter plasmid DNA (1 μg) and Renilla plasmid DNA (5 ng) were re-suspended in 10 μl of solution R. The incorporation tube was filled with 3 ml of solution E. The mixture containing plasmid DNA and cells was inserted into the microporator pipette station. HMC-1 cells were shocked using 1750 V/20 mA/plus width 1, and HL-60 cells were shocked using 1350V/35 mA/plus width 1. Plates were incubated for 24 h. The luciferase activities were determined using the Dual Luciferase Reporter Assay system (Promega). The pGL3-control (Promega) and the promoter-less pGL3-basic vectors were used as positive and negative controls, respectively. Each experiment was conducted in triplicate for each sample, and the results are expressed as means±s.d. of three independent experiments.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from HMC-1 and HL-60 cells and were subjected to the Bradford Assay to determine protein concentrations as described before.23 Extracts were quickly frozen and aliquots stored at −80 °C. The oligonucleotide sequences were 5′-TCTCCCAGTCGGCTCCTAAGG-3′ for −5993G and 5′-TCTCCCAGTCAGCTCCTAAGG-3′ for −5993A, respectively. Competitor oligonucleotides used were activator protein-1 (5′-TTCCGGCTGAGTCATCAAGCG-3′) and nuclear factor-κB (5′-AGTTGAGGGGACTTTCCCAGG-3′). The double-stranded oligonucleotides were annealed by incubation at 95 °C for 5 min followed by successive gradual cooling to 37 °C. The double-stranded DNA probes were radiolabeled at their 5′ ends with DNA polymerase I (Klenow; Promega) and [α-32P]CTP. Reaction mixtures of 10 μl were prepared containing 5 μg of nuclear protein, 10 mM l−1 HEPES (pH 7.9), 250 ng poly(dI-dC), 100 ng bovine serum albumin, 1 mM l−1 MgCl2, 30 mM l−1 KCl, 1 mM l−1 dithiothreitol, 1 mM l−1 NaNO3, 1 mM l−1 NaF and 5% glycerol. These were preincubated on ice for 20 min before further 20 min of incubation in the presence of 0.2 pmol radiolabeled probe. The reaction mixtures were then subjected to electrophoresis on a native 6% polyacrylamide gel. In the competitor experiments, unlabeled blunt-ended competitor oligonucleotides were added to the binding reaction mixtures before addition of radiolabeled oligonucleotide probes. All gels were dried and radioactivity was detected the following day, using a FLA3000 scanner (Fuji Photo Film, Tokyo, Japan).
Statistical methods
The genotype frequency of each SNP was examined for a significant departure from the Hardy–Weinberg equilibrium using a χ2 test. Differences in genotype frequencies between the two groups were examined by means of a logistic regression analysis in co-dominant, dominant and recessive models after accounting for age and gender as covariables. Differences in the mean values of the phenotypic characteristics among the patients, according to their genotype, were compared using χ2 analysis for categorical variables, and independent t-tests for continuous variables. The dual luciferase assay was assessed using an independent t-test. All ECP, peripheral blood eosinophils, and total IgE data were log10-transformed before analysis for normal distribution. The P-values of <0.05 were considered to indicate significance. All statistical analyses were performed using SPSS, version 12.0 (SPSS, Chicago, IL, USA).
Results
Clinical characteristics of the study subjects
The patients with AERD were significantly older than both ATA and NC patients (P<0.001), as summarized in Table 1. Significant differences in prevalence of nasal polyps and atopy rate were found between the AERD and the ATA groups (P<0.001, respectively). The baseline FEV1% and methacholine PC20, the provocative concentration of methacholine required to cause a 20% fall in FEV1, levels were significantly lower in patients with AERD (P<0.001, respectively), whereas the percentage decrease in FEV1 after aspirin provocation was significantly greater in AERD patients than in ATA patients (P<0.001). The presence of serum-specific IgE to staphylococcal superantigens was not significantly different in AERD and ATA patients.
Genotype frequencies of IL5RA gene with AERD phenotypes
The genotype distributions of three polymorphisms did not depart significantly from Hardy–Weinberg equilibrium (P>0.05). Genotype frequencies of the three SNPs in IL5RA were not significantly different in the AERD, ATA and NC groups (Table 2).
AERD patients with the −5993AA genotype had higher presence of serum-specific IgE to staphylococcal enterotoxin A (P=0.008) than those with the −5993GG/GA genotype (Table 3), whereas the prevalence of serum-specific IgE to staphylococcal enterotoxin B and toxic shock syndrome toxin 1 was not significantly different according to the genotypes (P>0.05). No significant associations were found in atopic status, chronic rhinosinusitis, nasal polyp, total IgE level, ECP and peripheral eosinophil counts according to the −5993A>G polymorphism in AERD patients (Table 3). Also, no significant associations were found between the clinical parameters and the −5567C>G and −5091G>A polymorphisms in AERD patients (P>0.05, respectively).
Effect of the IL5RA -5993G>A polymorphism on transcriptional activity
To determine whether the −5993G>A polymorphism was associated with altered promoter activity, we used a luciferase reporter assay to compare the reporter activity of plasmids that contained either −5993G or −5993A in the IL5RA in HMC-1 and HL-60 cells. The constructs containing −5993A exhibited significantly greater luciferase activity compared with those containing −5993G in HMC-1 and HL-60 cells (P=0.030 and P=0.013, respectively; Figures 1b and c, respectively).

Effect of the interleukin-5 receptor α (IL5RA) −5993G>A polymorphism on transcriptional activity. Plasmid constructs used for transfection (a). Transfection of HMC-1 (human mast cell line) cells (b). Transfection of human promyelocytic leukemia (HL-60) cells (c). A luciferase activity assay was performed in three independent experiments (total, n=9). Relative luciferase activity values are shown as means±s.d. The P-value was determined using the independent t-test.
Next, we examined the allelic differences of −5993G>A in the binding of nuclear proteins by EMSA. The signal intensity of the DNA–protein complex derived from the −5993A allele was higher than that from the −5993G allele in the presence of HMC-1 (Figure 2a) and HL-60 (Figure 2b) nuclear extracts. This complex was not visible in the presence of the nonlabeled −5993A probe as a competitor and was diminished by excess amounts of other nonlabeled competitor probes.

Electrophoretic mobility shift assay (EMSA) findings for the interleukin-5 receptor α (IL5RA) −5993G>A polymorphism. Differential binding of a nuclear protein to −5993G/A alleles in HMC-1 (human mast cell line) cells (a). Differential binding of a nuclear protein to −5993G/A alleles in human promyelocytic leukemia (HL−60) cells (b). The same results were obtained in at least three independent experiments.
Discussion
Genetic associations of IL5RA have been reported in patients with bronchial asthma and atopic dermatitis in a Korean population.11, 12, 13 In 2005, Cheong et al.11 identified 22 polymorphic sites within IL5RA containing exons and flanking region, in a Korean population. When a further 10 common polymorphisms were considered, they determined the A allele at the promoter polymorphism −5993G>A, which is considered to be a risk allele for adult asthmatic patients.11 Flow cytometry showed that eosinophils from adult asthmatic patients carrying the A allele at −5091G>A exhibited higher IL5RA protein expression.12 To our knowledge, there have been no published reports of the genetic association of IL5RA polymorphisms with the pathogenic mechanism of AERD. In the present study, we selected and genotyped three SNPs within IL5RA: −5993G>A, −5567C>G and −5091G>A. They were not in strong linkage disequilibrium (r2<0.21), and we therefore hypothesize that these SNPs may act independently to contribute to eosinophil activation in AERD.
Eosinophilic infiltration in the upper and lower airway mucosa is the common feature in AERD,3, 24 which is regulated by the IL-5 cytokine through binding with the receptor α-subunit (IL5RA).25 Increased IL5RA protein and mRNA levels have been reported in various allergic disorders and this is in correlation with an increase in blood eosinophils.26, 27, 28 In the present study, although no significant differences were found in the genotype frequencies of the three SNPs within IL5RA between the AERD and ATA patients, AERD patients carrying −5993A allele had higher presence of serum-specific IgE. Moreover, a luciferase reporter assay using human promyelocytic leukemia cells, which differentiate toward the eosinophils, showed that the IL5RA −5993A allele may increase expression of the IL5RA. These findings suggest that the human IL5RA promoter polymorphism at −5993G>A may contribute to eosinophil activation via increased expression of IL5RA.
Recent studies have provided evidence indicating that staphylococcal superantigens could induce IL-5 synthesis and promote eosinophil activation, and IL-5 is reported to induce cysteinyl leukotriene synthesis, which is a major eicosanoid involved in the pathogenic mechanism of AERD.29, 30 Similarly, eicosanoids have been reported to be upregulated in nasal polyp tissue along with the presence of specific IgE to the staphylococcal superantigen. Moreover, the concentration of eicosanoid products is correlated to eosinophilic inflammation markers and levels of specific IgE to superantigens, indicating that upregulation of eicosanoid synthesis is related to the inflammatory reaction induced by staphylococcal superantigens in AERD.31 Staphylococcal superantigens have the potential to stimulate polyclonal T- and B-cell proliferation, induce a class switching to IgE, produce allergen-specific IgE and release inflammatory cytokines and mediators to increase airway inflammation and bronchial hyperresponsiveness, all of which directly affect disease severity.17, 32 We also observed increased levels of specific IgE to staphylococcal enterotoxins A and B in nasal polyp tissue homogenates obtained from the AERD patients, in which specific IgE was also present along with higher levels of ECP and IL-5.4 In this study, AERD patients carrying IL5RA −5993A allele had higher prevalence of staphylococcal enterotoxin A-specific IgE, indicating that the specific IgE responses to staphylococcal superantigens could induce IL-5 release from T helper type 2 cells, and activate eosinophils via IL5RA in AERD patients, which might be heightened by the increased expression of IL5RA in patients carrying the −5993A allele.
Although IL5RA is particularly important in the regulation of eosinophils, expression of functional IL5RA on mast cells has been reported.33, 34 In vitro, we demonstrated that the IL5RA −5993G>A polymorphism had a direct effect on basal transcription rates of the IL5RA gene in human mast cells. The constructs containing the −5993A allele showed higher promoter activity than the construct with −5993G in the HMC-1 cells. In addition, the EMSA findings demonstrated that −5993A-specific DNA binding was more pronounced and may be more important in the transcriptional regulation of the IL5RA gene, compared with −5993G. As well as eosinophil activation, the functional variant of IL5RA −5993G>A may promote mast cell activation to release inflammatory cytokines and mediators, as mast cell activation was observed in inflammation of the upper and lower airways of AERD patients.35, 36 Specific IgE responses to staphylococcal superantigens may stimulate the production of IL-5 and influence local immunoglobulin synthesis, which contributes to mast cell activation.37, 38 Given our data, we also suggest that IL5RA promoter polymorphism at −5993G>A may affect mast cell activation in AERD patients in association with staphylococcal superantigens. AERD patients tended to present more severe clinical phenotype, and anti-IL-5 antibody therapy could reduce asthma exacerbation in severe asthma patients with eosinophilic dominant type.39, 40 The present findings may provide further support for the concept that drug response to anti-IL-5 or anti-IL-5 receptor antibody in AERD patients may be different according to this functional genetic polymorphism.
In conclusion, the results of this study suggested that a functional polymorphism in IL5RA may contribute to eosinophil and mast cell activation along with specific IgE responses to staphylococcal enterotoxin A in AERD patients.
References
Mascia K, Haselkorn T, Deniz YM, Miller DP, Bleecker ER, Borish L . Aspirin sensitivity and severity of asthma: evidence for irreversible airway obstruction in patients with severe or difficult-to-treat asthma. J Allergy Clin Immunol 2005; 116: 970–975.
Chang HS, Park JS, Jang AS, Park SW, Uh ST, Kim YH et al. Diagnostic value of clinical parameters in the prediction of aspirin-exacerbated respiratory disease in asthma. Allergy Asthma Immunol Res 2011; 3: 256–264.
Nasser SM, Pfister R, Christie PE, Sousa AR, Barker J, Schmitz-Schumann M et al. Inflammatory cell populations in bronchial biopsies from aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med 1996; 153: 90–96.
Suh YJ, Yoon SH, Sampson AP, Kim HJ, Kim SH, Nahm DH et al. Specific immunoglobulin E for staphylococcal enterotoxins in nasal polyps from patients with aspirin-intolerant asthma. Clin Exp Allergy 2004; 34: 1270–1275.
Olze H, Förster U, Zuberbier T, Morawietz L, Luger EO . Eosinophilic nasal polyps are a rich source of eotaxin, eotaxin-2 and eotaxin-3. Rhinology 2006; 44: 145–150.
Sladek K, Dworski R, Soja J, Sheller JR, Nizankowska E, Oates JA et al. Eicosanoids in bronchoalveolar lavage fluid of aspirin-intolerant patients with asthma after aspirin challenge. Am J Respir Crit Care Med 1994; 149: 940–946.
Stevenson DD, Zuraw BL . Pathogenesis of aspirin-exacerbated respiratory disease. Clin Rev Allergy Immunol 2003; 24: 169–188.
Zagai U, Sköld CM, Trulson A, Venge P, Lundahl J . The effect of eosinophils on collagen gel contraction and implications for tissue remodelling. Clin Exp Immunol 2004; 135: 427–433.
Tavernier J, Devos R, Cornelis S, Tuypens T, Van der Heyden J, Fiers W et al. A human high affinity interleukin-5 receptor (IL5R) is composed of an IL5-specific alpha chain and a beta chain shared with the receptor for GM-CSF. Cell 1991; 66: 1175–1184.
Murata Y, Takaki S, Migita M, Kikuchi Y, Tominaga A, Takatsu K . Molecular cloning and expression of the human interleukin 5 receptor. J Exp Med 1992; 175: 341–351.
Cheong HS, Kim LH, Park BL, Choi YH, Park HS, Hong SJ et al. Association analysis of interleukin 5 receptor alpha subunit (IL5RA) polymorphisms and asthma. J Hum Genet 2005; 50: 628–634.
Lee JH, Chang HS, Kim JH, Park SM, Lee YM, Uh ST et al. Genetic effect of CCR3 and IL5RA gene polymorphisms on eosinophilia in asthmatic patients. J Allergy Clin Immunol 2007; 120: 1110–1117.
Namkung JH, Lee JE, Kim E, Cho HJ, Kim S, Shin ES et al. IL-5 and IL-5 receptor alpha polymorphisms are associated with atopic dermatitis in Koreans. Allergy 2007; 62: 934–942.
Yasruel Z, Humbert M, Kotsimbos TC, Ploysongsang Y, Minshall E, Durham SR et al. Membrane-bound and soluble alpha IL-5 receptor mRNA in the bronchial mucosa of atopic and nonatopic asthmatics. Am J Respir Crit Care Med 1997; 155: 1413–1418.
Busse WW, Katial R, Gossage D, Sari S, Wang B, Kolbeck R et al. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol 2010; 125: 1237–1244.
Palikhe NS, Kim SH, Jin HJ, Hwang EK, Nam YH, Park HS . Genetic mechanisms in aspirin-exacerbated respiratory disease. J Allergy 2012; 2012: 794890.
Kowalski ML, Cieślak M, Pérez-Novo CA, Makowska JS, Bachert C . Clinical and immunological determinants of severe/refractory asthma (SRA): association with Staphylococcal superantigen-specific IgE antibodies. Allergy 2011; 66: 32–38.
Bachert C, van Steen K, Zhang N, Holtappels G, Cattaert T, Maus B et al. Specific IgE against Staphylococcus aureus enterotoxins: an independent risk factor for asthma. J Allergy Clin Immunol 2012; 130: 376–381.
Lee JY, Kim HM, Ye YM, Bahn JW, Suh CH, Nahm D et al. Role of staphylococcal superantigen-specific IgE antibodies in aspirin-intolerant asthma. Allergy Asthma Proc 2006; 27: 341–346.
Park HS . Early and late onset asthmatic responses following lysine-aspirin inhalation in aspirin-sensitive asthmatic patients. Clin Exp Allergy 1995; 25: 38–40.
Palikhe NS, Kim SH, Kim JH, Losol P, Ye YM, Park HS . Role of Toll-like receptor 3 variants in aspirin-exacerbated respiratory disease. Allergy Asthma Immunol Res 2011; 3: 123–127.
Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR . Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989; 77: 51–59.
Palikhe NS, Sin HJ, Kim SH, Hwang EK, Ye YM, Park HS . Genetic variability of prostaglandin E2 receptor subtype EP4 gene in aspirin-intolerant chronic urticaria. J Hum Genet 2012; 57: 494–499.
Lewandowska-Polak A, Jedrzejczak-Czechowicz M, Makowska JS, Jarzebska M, Jankowski A, Kowalski ML . Lack of association between aspirin-triggered 15-hydroxyeicosatetraenoic acid release and mast cell/eosinophil activation in nasal polyps from aspirin-sensitive patients. J Investig Allergol Clin Immunol 2011; 21: 507–513.
Kotsimbos AT, Q. Hamid . IL-5 and IL-5 receptor in asthma. Mem Inst Oswaldo Cruz 1997; 92: 75–91.
Gevaert P, Bachert C, Holtappels G, Novo CP, Van der Heyden J, Fransen L et al. Enhanced soluble interleukin-5 receptor alpha expression in nasal polyposis. Allergy 2003; 58: 371–379.
Gevaert P, Hellman C, Lundblad L, Lundahl J, Holtappels G, van Cauwenberge P et al. Differential expression of the interleukin 5 receptor alpha isoforms in blood and tissue eosinophils of nasal polyp patients. Allergy 2009; 64: 725–732.
Semic-Jusufagic A, Gevaert P, Bachert C, Murray C, Simpson A, Custovic A . Increased serum-soluble interleukin-5 receptor alpha level precedes the development of eczema in children. Pediatr Allergy Immunol 2010; 21: 1052–1058.
Barnes PJ . Intrinsic asthma: not so different from allergic asthma but driven by superantigens? Clin Exp Allergy 2009; 39: 1145–1151.
Cowburn AS, Holgate ST, Sampson AP . IL-5 increases expression of 5-lipoxygenase-activating protein and translocates 5-lipoxygenase to the nucleus in human blood eosinophils. J Immunol 1999; 163: 456–465.
Pérez-Novo CA, Claeys C, Van Zele T, Holtapples G, Van Cauwenberge P, Bachert C . Eicosanoid metabolism and eosinophilic inflammation in nasal polyp patients with immune response to Staphylococcus aureus enterotoxins. Am J Rhinol 2006; 20: 456–460.
Wenzel SE, Busse WW . Severe asthma: lessons from the Severe Asthma Research Program. J Allergy Clin Immunol 2007; 119: 14–21.
Wilson TM, Maric I, Shukla J, Brown M, Santos C, Simakova O et al. IL-5 receptor alpha levels in patients with marked eosinophilia or mastocytosis. J Allergy Clin Immunol 2011; 128: 1086–1092.
Dahl C, Hoffmann HJ, Saito H, Schiotz PO . Human mast cells express receptors for IL-3, IL-5 and GM-CSF; a partial map of receptors on human mast cells cultured in vitro. Allergy 2004; 59: 1087–1096.
Ochi H, De Jesus NH, Hsieh FH, Austen KF, JA. Boyce . IL-4 and -5 prime human mast cells for different profiles of IgE-dependent cytokine production. Proc Natl Acad Sci USA 2000; 97: 10509–10513.
Moneret-Vautrin DA, Hsieh V, Wayoff M, Guyot JL, Mouton C, Maria Y . Nonallergic rhinitis with eosinophilia syndrome a precursor of the triad: nasal polyposis, intrinsic asthma, and intolerance to aspirin. Ann Allergy 1990; 64: 513–518.
Bachert C, Zhang N, Patou J, van Zele T, Gevaert P . Role of staphylococcal superantigens in upper airway disease. Curr Opinion Allergy Clin Immunol 2008; 8: 34–38.
Jabara HH, Geha RS . The superantigen toxic shock syndrome toxin-1 induces CD40 ligand expression and modulates IgE isotype switching. Int Immunol 1996; 8: 1503–1510.
Palikhe NS, Kim JH, Park HS . Update on recent advances in the management of aspirin exacerbated respiratory disease. Yonsei Med J 2009; 50: 744–750.
Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 2012; 380: 651–659.
Acknowledgements
This work was supported by a grant from the Korean Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (A111218-11-PG01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Losol, P., Kim, SH., Seob Shin, Y. et al. A genetic effect of IL-5 receptor α polymorphism in patients with aspirin-exacerbated respiratory disease. Exp Mol Med 45, e14 (2013). https://doi.org/10.1038/emm.2013.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.24
Keywords
This article is cited by
-
Aspirin exacerbated respiratory disease (AERD): molecular and cellular diagnostic & prognostic approaches
Molecular Biology Reports (2021)
-
Efficacy of benralizumab for patients with severe eosinophilic asthma: a retrospective, real-life study
BMC Pulmonary Medicine (2020)
