
Abstract
Intrahepatic cholangiocarcinoma (ICC), a malignant tumor derived from the intrahepatic bile duct epithelium, has a poor prognosis and is refractory to conventional chemotherapy and radiation therapy. Thus, there is an urgent need to develop new effective therapeutic strategies for this disease. We previously found that L1 cell adhesion molecule (L1CAM) plays an important role in tumor progression of ICC, and we generated a murine mAb, A10-A3 (IgG1), that binds to the Ig1 domain of L1CAM. In the present study, we further characterized A10-A3, constructed a chimeric A10-A3 antibody (cA10-A3) containing the constant regions of human IgG1, and evaluated the therapeutic potential in a human ICC xenograft nude mice model. The affinities (KD) of A10-A3 and cA10-A3 for soluble L1CAM were 1.8 nM and 1.9 nM, respectively, as determined by competition ELISA. A10-A3 inhibited L1CAM homophilic binding and was slowly internalized into the tumor cells, but it did not significantly inhibit proliferation of ICC cells in vitro. cA10-A3 mediated antibody-dependent cell-mediated cytotoxicity in vitro and displayed anti-tumor activity in the ICC animal model. These results suggest that the humanized A10-A3 antibody may have potential as an anticancer agent for the treatment of ICC.
Similar content being viewed by others


Introduction
Cholangiocarcinoma is a malignant tumor that arises from the bile duct epithelium. Cholangiocarcinoma is classified anatomically into intrahepatic cholangiocarcinoma (ICC) and extrahepatic cholangiocarcinoma (ECC) that have different risk factors, pathogeneses, and clinical features due to the distinct development and differentiation of intrahepatic and extrahepatic bile duct epithelium (Nakeeb et al., 1966; Shiojiri, 1997; Shaib et al., 2007). ICC occurs at a higher incidence in Southeast Asia than in Europe and North America, but the incidence and mortality rates are increasing worldwide (Nakeeb et al., 1966; Patel, 2001; Taylor-Robinson et al., 2001). Since cholangiocarcinoma is refractory to conventional chemotherapy and radiation treatment (Sirica, 2005), complete surgical resection is currently the only treatment and cure. However, because of a lack of early diagnosis, most patients have occult metastasis or advanced local disease on clinical presentation (Lazaridis and Gores, 2005). Moreover, prognosis of cholangiocarcinoma is very poor (Khan et al., 2005). Thus, new effective therapeutic strategies for this disease are urgently needed.
The L1 cell adhesion molecule (L1CAM) is a member of the immunoglobulin (Ig) superfamily; it is a 200 to 220-kDa transmembrane glycoprotein consisting of six Ig-like domains followed by five fibronectin (Fn)-type III repeats, a transmembrane domain, and a short cytoplasmic tail (Moos et al., 1988). L1CAM was first described as a neural cell adhesion molecule and has been shown to play key roles in the development of the nervous system, including cell adhesion, neurite outgrowth, axon guidance, neural cell migration, and myelination (Rathjen and Schachner, 1984; Brummendorf and Rathjen, 1995; Kaifi et al., 2006). L1CAM promotes cellular activities through L1 homophilic interaction, as well as heterophilic interaction with other neuronal members of the Ig superfamily, integrins, extracellular matrix proteins and cell surface receptors (reviewed in Haspel and Grumet, 2003). Recent reports have shown that L1CAM is aberrantly expressed in several different types of cancers, including colon carcinoma, ovarian and uterine carcinomas, malignant gliomas, recurrent neuroblastoma, cutaneous malignant melanoma, renal cell carcinoma, ECC and gallbladder carcinoma, and that its expression correlates with more advanced stages of tumor progression (Li et al., 2009; reviewed in Raveh et al., 2009; Choi et al., 2011). In addition, ectopic L1CAM expression in carcinoma cells enhances their migration, invasion, and tumorigenesis (Silletti et al., 2004; Gast et al., 2005; Gavert et al., 2005; Min et al., 2010). In addition to functioning as a cell surface adhesion molecule, the extracellular domain of L1CAM can be shed from the cell surface via proteolytic cleavage and can stimulate the migration and survival of tumor cells through autocrine/paracrine binding to integrins (Duczmal et al., 1997; Mechtersheimer et al., 2001; Voura et al., 2001; Gutwein et al., 2003). A monoclonal antibody (mAb) against L1CAM reportedly inhibits the growth and dissemination of ovarian carcinomas in nude mice (Arlt et al., 2006; Wolterink et al., 2010).
By immunizing mice with human ICC cell lines, we previously generated a murine mAb, A10-A3 (IgG1, κ), that specifically binds to human L1CAM (Min et al., 2010). Immunohistochemical analysis of ICC tumor tissues using A10-A3 revealed L1CAM expression in 40.5% of the ICC patients. A functional study of L1CAM suppression or overexpression in ICC tumor cells indicated that L1CAM plays an important role in tumor progression of ICC by promoting cell proliferation, migration, and survival (Min et al., 2010). These results suggested that L1CAM may serve as a therapeutic target in ICC and that anti-L1CAM mAb may have potential as diagnostic and therapeutic agents for the treatment of ICC. To characterize A10-A3, we performed epitope mapping, which showed that A10-A3 binds to the Ig1 domain of L1CAM (Wei et al., 2011). In the present study, we have further characterized A10-A3, constructed chimeric A10-A3 (cA10-A3), and evaluated its therapeutic potential in a human ICC xenograft nude mice model.
Results
Affinity determination of A10-A3
To determine the affinity of A10-A3 for L1CAM, hL1-ECD-S1 was produced and purified by affinity chromatography on anti-preS1 mAb KR127-coupled Sepharose columns (Maeng et al., 2000; Ryu et al., 2000). After its purity and integrity was confirmed by SDS-PAGE (Figure 1A, lane 2), the purified hL1-ECD-S1 protein was used as an antigen to determine the affinity of A10-A3 by an indirect ELISA followed by a competition ELISA (Figures 1B and 1C). The affinity (KD) of A10-A3 for hL1-ECD-S1 was 1.8 nM.

(A) Analysis of purified L1CAM proteins. Lane 1, purified hL1-ECD-Fc; Lane 2, hL1-ECD-S1; and Lane M, molecular weight standards. Proteins were subjected to 10% SDS-PAGE. Protein bands were visualized by Commassie blue staining. (B) Affinity determination of anti-L1CAM mAbs. Antigen-binding activities of A10-A3 (•) and cA10-A3 (▽) were determined by indirect ELISA. (C) Affinity determination of A10-A3. Antigen-binding affinities of A10-A3 (•) and chimeric A10-A3 (▽) were determined by competition ELISA. Inset represents a Klotz plot.
Internalization of A10-A3 into tumor cells
The anti-cancer therapeutic antibodies trastuzumab and cetuximab bind to Her2 and epidermal growth factor receptor (EGFR), respectively, and are internalized into target tumor cells where they down regulate receptor expression on the cell surface (Mendelsohn, 1997; Liang et al., 2003). To examine whether A10-A3 is internalized after binding to the cells, we incubated Choi-CK, SCK (Kim et al., 2008), or L1CAM-overexpressing SCK-L1 cells (Min et al., 2010) with A10-A3 for indicated time periods. Then the cells were labeled with an Alexa-488-conjugated secondary antibody, and analyzed by confocal microscopy. In 1 h, A10-A3 effectively bound to the plasma membrane of the cells, and it was internalized into endocytic vesicles of the SCK-LI cells at 6 h after the antibody treatment (Figure 2B). However, internalization of A10-A3 was not detected in Choi-CK and SCK cells (data not shown), probably because these cells express lower levels of L1CAM compared to SCK-L1 cells (Figure 2A).

(A) Flow cytometric analysis of ICC cells. Levels of L1CAM expression in ICC cells were analyzed by flow cytometry using A10-A3. Uncolored populations indicate A10-A3 staining; colored populations indicate control staining. (B) Confocal microscopic analysis of A10-A3 internalization. SCK-L1 cells were preincubated with A10-A3 (20 µg/ml) for the indicated times. A10-A3 was detected using Alexa-488-conjugated goat anti-mouse IgG (green), and nucleus was counterstained with DAPI (blue). Merging of the two signals is shown at lower right panel for each time point.
Inhibition of L1CAM homophilic binding by A10-A3
It was recently demonstrated that the four N-terminal Ig-like domains (Ig1-4) mediate L1CAM homophilic binding in trans with approximately 100 nM of KD and that this promotes neurite outgrowth from human neurons (Gouveia et al., 2008). Additionally, Ig6 was shown to be involved in heterophilic binding with integrins (Haspel and Grumet, 2003). Since A10-A3 binds to the Ig1 domain with a high affinity, it may efficiently block L1CAM homophilic binding. To test this, a cell-free homophilic binding assay was established using purified hL1-ECD-S1 and hL1-ECD-Fc that was captured by anti-Fc antibodies immobilized on the wells (Figure 1A). As shown in Figure 3B, hL1-ECD-S1 bound to hL1-ECD-Fc, but did not bind to purified Ig1-Fc (Figure 3A, lane 2) or BSA. Subsequently, hL1-ECD-S1 was pre-incubated with different concentrations of A10-A3 and allowed to bind to hL1-ECD-Fc. A10-A3 inhibited the homophilic binding in a dose-dependent manner (Figure 3B).

(A) Analysis of purified L1CAM proteins. Lane 1, purified hL1-ECD-Fc; Lane 2, Ig1-Fc; Lane 3, Ig5-6/Fn1-5-Fc; and Lane M, molecular weight standards. Proteins were subjected to 10% SDS-PAGE. Protein bands were visualized by Commassie blue staining. (B) L1CAM homophilic binding assay; 2 µM hL1-ECD-S1 (hL1-S1) was incubated with purified hL1-ECD-Fc (hL1-Fc), Ig1-Fc, or BSA coated on each well. For the inhibition assay, 2 µM hL1-S1 was preincubated with A10-A3 (0, 1, 2, 5, 10, 20, or 50 µg) and allowed to bind to hL1-Fc captured by anti-Fc coated on the well. (C) Cell adhesion assay. SCK-L1 cells were incubated with purified hL1-Fc, Ig1-Fc, Ig5-6/Fn1-5-Fc, or BSA. For the inhibition assay, SCK-L1 cells were preincubated with mock or 10 µg of A10-A3, UJ127, or KR127 and allowed to bind to hL1-Fc coated on the well. Assays were performed three independent times in duplicate.
Next, a cell adhesion assay was performed by using SCK-L1 cells and hL1-ECD-Fc that was captured by anti-Fc antibodies immobilized on the wells. As shown in Figure 3B, SCK-L1 cells bound to hL1-ECD-Fc, but did not bind to Ig1-Fc or BSA. The cells also showed weak binding to purified Ig5-6/Fn1-5-Fc (Figure 3A, lane 3), which lacks Ig1-4 that mediate homophilic binding but contains Ig6 that can bind to integrins. The binding of Ig5-6/Fn1-5-Fc to the cells was approximately 25% that of hL1-ECD-Fc, indicating that L1CAM homophilic binding mediated by Ig1-4 is much stronger than L1CAM-integrin binding. For the inhibition study, cells were preincubated with A10-A3 or control antibody for 3 h and allowed to bind to hL1-ECD-Fc. A10-A3 efficiently inhibited the binding between the cells and hL1-ECD-Fc, decreasing the binding to a level that was similar to the binding observed between the cells and Ig5-6/Fn1-5-Fc. On the other hand, the UJ127 antibody, which binds to the Fn domain of L1CAM (Arlt et al., 2006) or KR127 as an isotype control, did not inhibit the binding of the cells to hL1-ECD. The results suggest that A10-A3 specifically inhibits L1CAM homophilic binding.
Finally, a proliferation assay was performed using Choi-CK, SCK, and SCK-L1 cells, as described in the materials and methods. A10-A3 hardly inhibited the proliferation of the cells (data not shown).
Construction and characterization of cA10-A3
Distinct subclasses of IgG display substantial differences in their ability to mediate effector functions (Nimmerjahn and Ravetch, 2005). The mouse IgG2a was most effective and much more efficient than the mouse IgG1 variant of the same mAb, while human IgG1 was most effective in ADCC and CDC (Nimmerjahn and Ravetch, 2006). With the aim of validating the therapeutic potential of A10-A3 in nude mice bearing ICC xenograft, we replaced mouse IgG1 with human IgG1 to construct cA10-A3. The resulting chimeric antibody was expressed in DHFR-deficient CHO cells. After selection in G418 and adaptation in a medium containing 20 nM MTX, the transfected cells were cultured in a protein-free medium and the chimeric antibody secreted in the culture supernatant was purified by affinity chromatography on a Protein A column. The purified cA10-A3 was subjected to competition ELISA for affinity determination; the affinity (KD) of cA10-A3 was 1.9 nM, similar to that of A10-A3 (Figures 1B and 1C).
ADCC activity of cA10-A3
To evaluate whether cA10-A3 mediates antibody-dependent cell-mediated cytotoxicity (ADCC), a standard lactate dehydrogenase (LDH) assay was performed using SCK-L1 as target cells and human PBMC as effector cells. The SCK-L1 cells were pre-incubated with serially diluted cA10-A3 (0-10 µg/ml), then incubated with human peripheral blood mononuclear cells (PBMC) at a 30:1 ratio of effector:target (E:T) cells. The cA10-A3 was effective in inducing ADCC against SCK-L1 cells in a dose-dependent manner (Figure 4A), and the ADCC activity of antibody at 10 µg/ml concentration was increased with the ratio of E:T 60:1 (Figure 4B). However, the antibody did not induce ADCC against Choi-CK cells that express low level of L1CAM (data not shown). This may be because L1CAM expression in Choi-CK cells is too low to induce ADCC.

ADCC activity of cA10-A3. (A) ADCC assay using SCK-L1 (target cells) and PBMC (effector cells). The ratio of effector:target (E:T) was 30:1. (B) ADCC assay using 10 µg/ml cA10-A3 and a 30:1 or 60:1 ratio of E:T.
Inhibition of ICC tumor growth in nude mice by cA10-A3
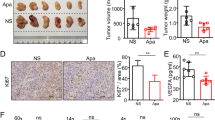
To evaluate the therapeutic efficacy in vivo, cA10-A3 or the isotype control palivizumab (Synagis, a humanized antibody to respiratory syncytial virus F protein) was i.v. injected three times a week for 28 days into nude mice bearing Choi-CK xenografts. The cA10-A3 inhibited the tumor growth in nude mice without affecting body weight (Figure 5). Since we observed that cA10-A3 did not induce ADCC against the Choi-CK cells in vitro, we examined whether the Choi-CK cells in the xenograft expressed a higher level of L1CAM compared to that of the cells cultured in vitro. Choi-CK cells were injected into nude mice and the tumor cells from the xenograft at day 7 post injection was prepared and subjected to flow cytometric analysis. The result showed that the level of L1CAM expression in the tumor cells from the Choi-CK xenograft was significantly higher than that in the Choi-CK cells cultured in vitro (Supplemental Data Figure 1), suggesting that the increased L1CAM expression level in the Choi-CK cells may be sufficient to induce ADCC in nude mice.

Inhibition of ICC tumor growth in nude mice by cA10-A3 treatment. (A) cA10-A3 antibody or the isotype control Synagis was intravenously injected at a dose of 10 mg/kg three times a week for 28 days in nude mice bearing Choi-CK xenograft. (B) Body weights of tumor-bearing mice.
Discussion
Cholangiocarcinoma is a malignant tumor with a poor prognosis and is refractory to conventional chemotherapy and radiation treatment. Thus, new effective therapeutic strategies for cholangiocarcinoma are urgently needed. L1CAM is aberrantly expressed in several types of tumors and plays important roles in tumor progression by enhancing cell proliferation, migration, and survival. Anti-L1CAM mAb has displayed therapeutic activity in a mouse model for ovarian carcinoma (Arlt et al., 2006; Wolterink et al., 2010). Previously, we found that L1CAM is involved in tumor progression of ICC, ECC, and gallbladder carcinoma; we generated a murine mAb, A10-A3 (IgG1) binding to the Ig1 domain of L1CAM (Li et al., 2009; Min et al., 2010; Choi et al., 2011; Jung et al., 2011; Wei et al., 2011). In the present study, we constructed the cA10-A3 containing the constant regions of human IgG1, confirmed its ADCC activity in vitro, and evaluated its therapeutic activity in a Choi-CK xenograft model. The cA10-A3 inhibited tumor growth in nude mice, suggesting potential for anti-L1CAM therapeutic antibodies as an anti-cancer agent for the ICC treatment.
The results of the internalization assay indicated that A10-A3 was internalized into endocytic vesicles, suggesting that the L1CAM bound by A10-A3 may be also internalized. However, the endocytic vesicles were not detected until 3 h after addition of the antibody and were clearly detected at 6 h. This suggests that internalization of A10-A3 proceeds slowly in the cells and thus may not substantially impact the ADCC activity of A10-A3. In contrast, a previous study using human epidermoid carcinoma cells showed that anti-EGFR mAb, mAb-225 was internalized into endocytic vesicles within 1 h (Sunada et al., 1986).
L1CAM plays an important role in tumor progression through homophilic binding and heterophilic interaction with other cell surface proteins, such as growth factor receptors and integrins. For example, the Ig6 domain of L1CAM contains an RGD motif that interacts with αvβ3 integrin; inhibition of this binding significantly reduces haptotactic migration of melanoma cells (Montgomery et al., 1996). In addition, L1CAM interacts with fibroblast growth factor receptor to confer enhanced cell motility and invasion of L1CAM-overexpressing ovarian cancer cells (Zecchini et al., 2008). However, it has not been clearly shown whether inhibition of L1CAM homophilic binding can influence tumor cell proliferation. In the present study, we observed that A10-A3 inhibited L1CAM homophilic binding, but did not significantly inhibit ICC cell proliferation in vitro. This indicates that blocking L1CAM homophilic binding is not sufficient to inhibit tumor cell proliferation, and suggests that L1CAM homophilic binding may play a minor role in proliferation of ICC cells. Considering this finding along with the fact that L1CAM overexpression enhanced proliferation of ICC cells (Min et al., 2010), it seems likely that heterophilic interaction of L1CAM with integrins and/or growth factor receptors or other indirect mechanism may have promoted the cell proliferation. We found that EGFR, hepatocyte growth factor receptor, and integrins (αvβ3, αvβ5, and β1) are expressed on the surface of ICC cells (Yoon et al., 2012). It will be interesting to study the exact mechanism of heterophilic interactions between L1CAM and these molecules, and how these interactions influence cellular functions in ICC cells. It might also be valuable to generate anti-L1CAM antibodies that can inhibit these heterophilic interactions to evaluate their therapeutic potential for the development of more effective anticancer antibodies.
Taken together, our results suggest that cA10-A3 inhibits L1CAM homophilic binding and mediates ADCC, while it is internalized into the tumor cells slowly. The cA10-A3 displayed anti-tumor activity in a human ICC xenograft nude mice model. Therefore, a humanized A10-A3 antibody may be used as an anticancer agent for the treatment of ICC.
Methods
Cell culture
SCK and Choi-CK cell lines, established from tumor samples from Korean patients with ICC (Yoo et al., 2009), L1CAM-overexpressing SCK (SCK-L1) cells (Min et al., 2010), and human embryonic kidney HEK293 cells were grown in DMEM (Invitrogen) with 10% fetal bovine serum (FBS; Hyclone) in 5% CO2, 37℃ humidified incubator. The dihydrofolate reductase (DHFR)-deficient Chinese hamster ovary (CHO) cell line, DG44 were grown in DMEM/F12 (Invitrogen) supplemented with hypoxanthine (10 mg/L), thymidine (10 mg/L), glycine (50 mg/L), glutamine (587 mg/L), glucose (4.5 mg/L), 10% fetal bovine serum, and Antibiotics-Antimycotics (Invitrogen).
Expression and production of the extracellular domains of human L1CAM
The cDNA (L1 cDNA) isolated from Choi-CK cells (Min et al., 2010) was used as template for PCR. The cDNA encoding the hL1-ECD (amino acids, aa 1-1082) was synthesized by PCR from the L1 cDNA using pfu turbo polymerase (Stratagene) and the primers (forward primer, 5'-ATG GTCGTGGCGCTGCGGTAC-3'; reverse primer, 5'-TGGAG ACTGTTCCGTGATGAC-3'). PCR reactions were performed in a thermocycler (MJ research) with the following cycling parameters: denaturation at 94℃ for 5 min; then, 25 repetitive cycles of 95℃ for 45 seconds, annealing at 56℃ for 45 seconds, and elongation at 72℃ for 4 min. The final extension was performed at 72℃ for 7 min followed by rapid cooling at 4℃. The resulting PCR product was cloned into pCR2.1-TOPO (Invitrogen) and sequenced by the DNA ABI 377 automated sequencer. Results were analyzed using the blast search tool in NCBI (http://www.ncbi.nlm.nih.gov/).
For the expression of the hL1-ECD fused to the preS1 tag (Oh et al., 2003) or the Fc of human immunoglobulin IgG1, the hL1-ECD cDNA was subcloned into the EcoRI-XhoI sites of pJK-dhfr2 (Aprogen, Korea) containing the preS1 tag or pJK-dhfr2-Fc (Aprogen, Korea) to construct pJK-dhfr2-hL1-ECD-S1 or pJK-dhfr2-hL1-ECD-Fc, respectively. For the expression of the Ig1 or Ig5-6/Fn1-5 fused to the Fc, the cDNA encoding the Ig1 (aa 1-126) or Ig5-6/Fn1-5 (aa 396-1082) was synthesized by PCR and subcloned into pJK-dhfr2-Fc to construct pJK-dhfr2-Ig1-Fc or pJK-dhfr2-Ig5-6/Fn1-5-Fc, respectively. The resulting expression plasmid was separately introduced into human embryonic kidney (HEK) 293T cells using Lipofectamin® 2000 (Invitrogen) according to the manufacturer's instructions. After the transfected cells were cultured in protein-free medium CD293 (Gibco/BRL), the culture supernatants were subjected to affinity chromatography on CNBr-Sepharose 4B FF resin (Amersham Pharmacia Biotech) conjugated with anti-preS1 mAb KR127 (Maeng et al., 2000; Ryu et al., 2000) or Protein A-agarose column (Upstate Biotechnology). The purified protein was quantified using NANO-DROP 2000 (Thermo).
Indirect ELISA
Microtiter wells were coated with the purified hL1-ECD-S1 (100 ng) diluted in 50 mM sodium carbonate buffer (pH 9.6) at 4℃ overnight and blocked with skim milk (2%) in PBS. The plates were washed four times with PBST (PBS containing 0.05% Tween 20) between steps. A10-A3 or cA10-A3 (0-100 ng) was added to each well, then anti-mouse IgG-HRP or anti-human Fc-HRP (Thermo, 1:10,000 v/v) was added to the wells. All incubations were carried out at 37℃ for 1 h. Color was developed with OptEIA TMB Substrate (BD), and the absorbance was measured at 450 nm in a microtiter plate reader (Versa max, Molecular Devices).
Competition ELISA
A solution containing 3.165 ng of purified A10-A3 or cA10-A3 and various concentrations (5 × 10-13 - 5 × 10-8 M) of the purified hL1-ECD-S1 protein was preincubated at 37℃ for 3 h, then the mixture was added to each well coated with 100 ng of hL1-ECD-S1 and subjected to an indirect ELISA. Equilibrium dissociation constant (KD) was calculated from a Klotz plot (Friguet et al., 1985).
Flow cytometry
Cells were incubated with A10-A3 for 60 min at 4℃. After washing twice with PBA, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-mouse immunoglobulin (Ig) (BD Pharmingen) for 30 min at 4℃. Propidium iodide (PI)-negative cells were analyzed for antibody binding using FACSCalibur (BD Immunocytometry System) and Cell Quest software (BD Immunocytometry System).
Cell internalization assay
SCK-L1 cells (1.5 × 104) were seeded in each well of cover glass bottom dishes (SPL, Korea) with growth medium (0.1 mL) and incubated at 37℃ for 24 h. The cells were incubated with A10-A3 (20 µg/ml) at 37℃ for 1 h, 3 h, 6 h and 12 h, washed twice with PBS, fixed with 2% paraformaldehyde, and permeabilized in PBS containing 0.2% Triton X-100 for 30 min on ice. The cells were blocked in PBS containing 2% BSA and 1% horse serum for 1 h at room temperature, and incubated with an Alexa-488-conjugated goat anti-mouse IgG (1:500) for 1 h at room temperature with gentle shaking. The cells were washed 6 times and stained with 4,6-diamidino-2-phenylindole (DAPI, 0.25 µg/ml) in PBS containing 0.1% Triton X-100 for 20 min at room temperature with gentle shaking. Fluorescence images were visualized on LSM 510 META laser scanning confocal imaging system (Carl Zeiss, Germany). Alexa 488-labeled A10-A3 was excited at 488 nm, and emission was recorded at 505-550 nm. DAPI was excited at 405 nm, and emission was recorded at 420-480 nm.
L1CAM homophilic binding assay
Microtiter plates were coated with anti-human IgG (Fc specific; Pierce) at a concentration of 25 µg/mL for 1 h at 37℃, then purified hL1-ECD-Fc or Ig1-Fc was added to each well at 2 µM concentration and incubated at 37℃ for 2 h. Control wells were coated with 2 µM BSA. After washing with PBST, hL1-ECD-S1 (2 µM in PBS containing 1% BSA) was added to each well and incubated at 37℃ for 3 h. To examine whether A10-A3 inhibits L1CAM homophilic binding, the hL1-ECD-S1 was preincubated with A10-A3 (0-50 µg) at 37℃ for 3 h, then the mixture was added to each well containing hL1-ECD-Fc and incubated at 37℃ for 3 h. To detect the bound hL1-ECD-S1, KR127 was conjugated with HRP using EZ-Link® Plus Activated Peroxidase (Thermo), and the resulting KR127-HRP conjugate (1:1,000 v/v) was added to each well and incubated at 37℃ for 1 h. Color was developed with OptEIA TMB Substrate, and the absorbance was measured at 450 nm in a microtiter plate reader. Data are shown as means of triplicates ± SD.
Cell adhesion assay
SCK-L1 cells (2 × 104) in DMEM were preincubated with 10 µg /ml of A10-A3, UJ127 (Abcam), or KR127 at 37℃ for 3 h and incubated with hL1-ECD-Fc, Ig5-6/Fn1-5-Fc, or Ig1-Fc that had been captured by anti-human IgG at 37℃ for 3 h. After washing with PBS three times, the attached cells were measured using a WST1 assay kit (Roche). Absorbance was measured at 450 nm in a microtiter plate reader. Data are shown as means of triplicates ± SD.
Cell proliferation assay
Cells (2 × 105) were seeded in 6-well dishes (Falcon, Milian SA, Geneva, Switzerland) in 3 ml medium containing 10% FBS. After 12 h, when cells had attached to the plates, antibody (10 µg/ml) or PBS was added. After 72 h, cells were detached in PBS containing 1 mM EDTA, washed twice in PBS, and counted in a hemocytometer.
Construction and production of chimeric antibody
For the cloning of the cDNAs encoding the heavy chain variable region (VH) or light chain variable region (VL) of A10-A3 (IgG1, κ), total RNAs were isolated from A10-A3 hybridoma (Min et al., 2010) and the cDNAs encoding the variable regions were synthesized by polymerase chain reaction (PCR) using the 5'-primers specific for the VH or VL of murine antibodies (Kim et al., 2008) and the 3'-primers specific for the mouse Cγ1 or Cκ. After the cDNA synthesis, the PCR products were subcloned into pBluescript SK (+). The three clones of the VH or VL cDNA were sequenced. To determine the N-terminal sequences of the heavy and light chains of A10-A3, each chain was isolated after SDS-PAGE and subjected to N-terminal sequencing. For the expression of cA10-A3, the cDNAs encoding the VH or VL with cognate leader sequence were fused to human Cγ1 or Cκ by subcloning into the EcoRI-ApaI or HindIII-BsiWI site, respectively, of pdCMV-dhfrC derived from pdCMV-dhfr-AKA (Yoon et al., 2006). The resulting expression plasmid, pdCMV-dhfrC-cA10A3 was introduced into DG44 cells using Lipofectamine® 2000 (Invitrogen) according to the manufacturer's protocol. Stably transfected cell lines were selected in a medium containing G418 (550 µg/ml) and subsequently subjected to MTX selection for gene amplification, as described previously (Yoon et al., 2006). A recombinant CHO cell line secreting the chimeric antibody was grown in serum-free medium (CHO-S-SFMII, GIBCO/BRL), and the culture supernatant was subjected to affinity chromatography on Protein A-Sepharose 4B column (GE Lifesciences) according to the supplier's instruction. The purity and integrity of the purified antibody were assessed by 2100 Bioanalyzer (Agilent Technologies). For the quantification of the purified antibody, the optical density of 1.43 at 280 nm was taken for the protein concentration of 1 mg/ml.
Antibody-dependent cell-mediated cytotoxicity (ADCC) assay
PBMC as effector cells were isolated from blood of three healthy donors by density gradient centrifugation using Histopaque (Sigma-Aldrich). SCK-L1 as target cells (5 × 103) were plated into each well of a 96-well plate containing phenol red free RPMI-1640 medium (50 µl) with 1% FBS. Then, serially diluted antibody solution (25 µl) was added to the target cells and incubated for 15 min in 5% CO2, 37℃ humidified incubator. After 15 min, the target cells were incubated with the ratio of E:T 30:1 or 60:1 of freshly prepared PBMC (25 µl) for 4 h. The cytotoxicity was determined by measuring the LDH activity released from the cytosol of damaged cells using Cytotoxicity Detection Kit PLUS (LDH) (Roche Applied Science, Germany). Spontaneous release was determined by incubating target cells with antibody in RPMI medium containing 1% FBS, and maximum release was determined by complete lysis of target cells in RPMI-1640 medium containing 1% FBS with the cell lysis solution provided in the kit. Nonspecific release was determined by incubating target cells with PBMC in RPMI medium containing 1% FBS. The percentage of specific lysis was calculated as [specific lysis (%) = (specific release-nonspecific release) / (maximum release-spontaneous release) × 100], as described previously (Horton et al., 2008). Data are expressed as the mean of triplicate wells.
Xenograft nude mice assay
Nude mice (BALB/c Slc-nu, 5 weeks old) were obtained from Japan SLC, Inc (Japan). Mice were housed under specific pathogen free conditions for 7 days in accordance with the guidelines of the Animal Care Committee at Biotoxtech Co., Ltd (Ochang, Korea). Choi-Ck tumor tissue (8 mm3) was inoculated s.c. into the back of each mouse. After 7 days, when tumor volume reached 65-93 mm3 (n = 10 per group), cA10-A3 or Synagis as an isotype control at a dose of 10 mg/kg was injected intravenously three times per week for 28 days. Tumor growth was monitored biweekly by measuring the length and width of the tumor with a caliper and calculating tumor volume on the basis of the following formula: volume = 0.523Lw2, where L is length and W is width. On day 40, mice were euthanized. To determine the toxicity to the animals, the body weight of the animals was measured twice a week.
Statistical analysis
Data are presented as mean ± SD. and statistical comparisons between groups were performed using one-way ANOVA followed by Student's t test. A value of P < 0.05 was considered significant.
Abbreviations
- ADCC:
-
antibody dependent cell-mediated cytotoxicity
- cA10-A3:
-
chimeric A10-A3
- CDC:
-
complement dependent cytotoxicity
- CHO:
-
Chinese hamster ovary
- CNBr:
-
cyanogens bromide
- dhfr:
-
dihydrofolate reductase
- ECC:
-
extrahepatic cholangiocarcinoma
- ECD:
-
extracellular domains
- Fc:
-
fragment crystallizable region
- Fn:
-
fibronectin
- ICC:
-
intrahepatic cholangiocarcinomav
- L1CAM:
-
L1 cell adhesion molecule
- LDH:
-
lactate dehydrogenase
- VH:
-
heavy chain variable region
- VL:
-
light chain variable region
References
Arlt MJ, Novak-Hofer I, Gast D, Gschwend V, Moldenhauer G, Grunberg J, Honer M, Schubiger PA, Altevogt P, Kruger A . Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment . Cancer Res 2006 ; 66 : 936 - 943
Brummendorf T, Rathjen FG . Cell adhesion molecules 1: immunoglobulin superfamily . Protein Profile 1995 ; 2 : 963 - 1108
Choi SY, Jo YS, Huang SM, Liang ZL, Min JK, Hong HJ, Kim JM . L1 cell adhesion molecule as a novel independent poor prognostic factor in gallbladder carcinoma . Hum Pathol 2011 ; 42 : 1476 - 1483
Duczmal A, Schollhammer S, Katich S, Ebeling O, Schwartz-Albiez R, Altevogt P . The L1 adhesion molecule supports alpha v beta 3-mediated migration of human tumor cells and activated T lymphocytes . Biochem Biophys Res Commun 1997 ; 232 : 236 - 239
Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME . Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay . J Immunol Methods 1985 ; 77 : 305 - 319
Gast D, Riedle S, Schabath H, Schlich S, Schneider A, Issa Y, Stoeck A, Fogel M, Joumaa S, Wenger T, Herr I, Gutwein P, Altevogt P . L1 augments cell migration and tumor growth but not beta3 integrin expression in ovarian carcinomas . Int J Cancer 2005 ; 115 : 658 - 665
Gavert N, Conacci-Sorrell M, Gast D, Schneider A, Altevogt P, Brabletz T, Ben-Ze'ev A . L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers . J Cell Biol 2005 ; 168 : 633 - 642
Gouveia RM, Gomes CM, Sousa M, Alves PM, Costa J . Kinetic analysis of L1 homophilic interaction: role of the first four immunoglobulin domains and implications on binding mechanism . J Biol Chem 2008 ; 283 : 28038 - 28047
Gutwein P, Mechtersheimer S, Riedle S, Stoeck A, Gast D, Joumaa S, Zentgraf H, Fogel M, Altevogt DP . ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles . FASEB J 2003 ; 17 : 292 - 294
Haspel J, Grumet M . The L1CAM extracellular region: a multi-domain protein with modular and cooperative binding modes . Front Biosci 2003 ; 8 : s1210 - s1225
Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, Richards JO, Vostiar I, Joyce PF, Repp R, Desjarlais JR, Zhukovsky EA . Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia . Cancer Res 2008 ; 68 : 8049 - 8057
Jung J, Son YS, Park H, Jeon SK, Lee JW, Choi SY, Kim JM, Kwon YG, Hong HJ, Min JK . The cell adhesion molecule L1 promotes gallbladder carcinoma progression in vitro and in vivo . Oncol Rep 2011 ; 25 : 945 - 952
Kaifi JT, Heidtmann S, Schurr PG, Reichelt U, Mann O, Yekebas EF, Wachowiak R, Strate T, Schachner M, Izbicki JR . Absence of L1 in pancreatic masses distinguishes adenocarcinomas from poorly differentiated neuroendocrine carcinomas . Anticancer Res 2006 ; 26 : 1167 - 1170
Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD . Cholangiocarcinoma . Lancet 2005 ; 366 : 1303 - 1314
Kim SJ, Jang MH, Ahn HJ, Kim JH, Lim JH, Ryu CJ, Lim NK, Kim KS, Park MJ, Park I, Hong HJ . Selection of an affinity-matured antibody against a defined epitope by phage display of an immune antibody library . J Immunol Methods 2008 ; 329 : 176 - 183
Lazaridis KN, Gores GJ . Cholangiocarcinoma . Gastroenterology 2005 ; 128 : 1655 - 1667
Li S, Jo YS, Lee JH, Min JK, Lee ES, Park T, Kim JM, Hong HJ . L1 cell adhesion molecule is a novel independent poor prognostic factor of extrahepatic cholangiocarcinoma . Clin Cancer Res 2009 ; 15 : 7345 - 7351
Liang K, Lu Y, Jin W, Ang KK, Milas L, Fan Z . Sensitization of breast cancer cells to radiation by trastuzumab . Mol Cancer Ther 2003 ; 2 : 1113 - 1120
Maeng CY, Ryu CJ, Gripon P, Guguen-Guillouzo C, Hong HJ . Fine mapping of virus-neutralizing epitopes on hepatitis B virus PreS1 . Virology 2000 ; 270 : 9 - 16
Mechtersheimer S, Gutwein P, Agmon-Levin N, Stoeck A, Oleszewski M, Riedle S, Postina R, Fahrenholz F, Fogel M, Lemmon V, Altevogt P . Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins . J Cell Biol 2001 ; 155 : 661 - 673
Mendelsohn J . Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy . Clin Cancer Res 1997 ; 3 : 2703 - 2707
Min JK, Kim JM, Li S, Lee JW, Yoon H, Ryu CJ, Jeon SH, Lee JH, Kim JY, Yoon HK, Lee YK, Kim BH, Son YS, Choi HS, Lim NK, Kim DG, Hong HJ . L1 cell adhesion molecule is a novel therapeutic target in intrahepatic cholangiocarcinoma . Clin Cancer Res 2010 ; 16 : 3571 - 3580
Montgomery AM, Becker JC, Siu CH, Lemmon VP, Cheresh DA, Pancook JD, Zhao X, Reisfeld RA . Human neural cell adhesion molecule L1 and rat homologue NILE are ligands for integrin alpha v beta 3 . J Cell Biol 1996 ; 132 : 475 - 485
Moos M, Tacke R, Scherer H, Teplow D, Fruh K, Schachner M . Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin . Nature 1988 ; 334 : 701 - 703
Nakeeb A, Pitt HA, Sohn TA, Coleman J, Abrams RA, Piantadosi S, Hruban RH, Lillemoe KD, Yeo CJ, Cameron JL . Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors . Ann Surg 1996 ; 224 : 463 - 473
Nimmerjahn F, Ravetch JV . Divergent immunoglobulin g subclass activity through selective Fc receptor binding . Science 2005 ; 310 : 1510 - 1512
Nimmerjahn F, Ravetch JV . Fcgamma receptors: old friends and new family members . Immunity 2006 ; 24 : 19 - 28
Oh MS, Kim KS, Jang YK, Maeng CY, Min SH, Jang MH, Yoon SO, Kim JH, Hong HJ . A new epitope tag from hepatitis B virus preS1 for immunodetection, localization and affinity purification of recombinant proteins . J Immunol Methods 2003 ; 283 : 77 - 89
Patel T . Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States . Hepatology 2001 ; 33 : 1353 - 1357
Rathjen FG, Schachner M . Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion . EMBO J 1984 ; 3 : 1 - 10
Raveh S, Gavert N, Ben-Ze'ev A . L1 cell adhesion molecule (L1CAM) in invasive tumors . Cancer Lett 2009 ; 282 : 137 - 145
Ryu CJ, Kim YK, Hur H, Kim HS, Oh JM, Kang YJ, Hong HJ . Mouse monoclonal antibodies to hepatitis B virus preS1 produced after immunization with recombinant preS1 peptide . Hybridoma 2000 ; 19 : 185 - 189
Shaib YH, El-Serag HB, Nooka AK, Thomas M, Brown TD, Patt YZ, Hassan MM . Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a hospital-based case-control study . Am J Gastroenterol 2007 ; 102 : 1016 - 1021
Shiojiri N . Development and differentiation of bile ducts in the mammalian liver . Microsc Res Tech 1997 ; 39 : 328 - 335
Silletti S, Yebra M, Perez B, Cirulli V, McMahon M, Montgomery AM . Extracellular signal-regulated kinase (ERK)-dependent gene expression contributes to L1 cell adhesion molecule-dependent motility and invasion . J Biol Chem 2004 ; 279 : 28880 - 28888
Sirica AE . Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy . Hepatology 2005 ; 41 : 5 - 15
Sunada H, Magun BE, Mendelsohn J, Macleod CL . Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation . Proc Natl Acad Sci USA 1986 ; 83 : 3825 - 3829
Taylor-Robinson SD, Toledano MB, Arora S, Keegan TJ, Hargreaves S, Beck A, Khan SA, Elliott P, Thomas HC . Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968-1998 . Gut 2001 ; 48 : 816 - 820
Voura EB, Ramjeesingh RA, Montgomery AM, Siu CH . Involvement of integrin alpha(v)beta(3) and cell adhesion molecule L1 in transendothelial migration of melanoma cells . Mol Biol Cell 2001 ; 12 : 2699 - 2710
Wei CH, Lee ES, Jeon JY, Heo YS, Kim SJ, Jeon YH, Kim KH, Hong HJ, Ryu SE . Structural mechanism of the antigen recognition by the L1 cell adhesion molecule antibody A10-A3 . FEBS Lett 2011 ; 585 : 153 - 158
Wolterink S, Moldenhauer G, Fogel M, Kiefel H, Pfeifer M, Luttgau S, Gouveia R, Costa J, Endell J, Moebius U, Altevogt P . Therapeutic antibodies to human L1CAM: functional characterization and application in a mouse model for ovarian carcinoma . Cancer Res 2010 ; 70 : 2504 - 2515
Yoo HJ, Yun BR, Kwon JH, Ahn HS, Seol WA, Lee MJ, Yu GR, Yu HC, Hong BH, Choi K, Kim DG . Genetic and expression alterations in association with the sarcomatous change of cholangiocarcinoma cells . Exp Mol Med 2009 ; 41 : 102 - 115
Yoon H, Min JK, Lee JW, Kim DG, Hong HJ . Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2 . Biochem Biophys Res Commun 2011 ; 405 : 333 - 337
Yoon H, Min JK, Lee DG, Kim DG, Koh SS, Hong HJ . L1 cell adhesion molecule and epidermal growth factor receptor activation confer cisplatin resistance in intrahepatic cholangiocarcinoma cells . Cancer Lett 2012 ; 316 : 70 - 76
Yoon SO, Lee TS, Kim SJ, Jang MH, Kang YJ, Park JH, Kim KS, Lee HS, Ryu CJ, Gonzales NR, Kashmiri SV, Lim SM, Choi CW, Hong HJ . Construction, affinity maturation, and biological characterization of an anti-tumor-associated glycoprotein-72 humanized antibody . J Biol Chem 2006 ; 281 : 6985 - 6992
Zecchini S, Bianchi M, Colombo N, Fasani R, Goisis G, Casadio C, Viale G, Liu J, Herlyn M, Godwin AK, Nuciforo PG, Cavallaro U . The differential role of L1 in ovarian carcinoma and normal ovarian surface epithelium . Cancer Res 2008 ; 68 : 1110 - 1118
Acknowledgements
This work was supported by a grant from Korea Biotech R&D Group of Next-generation growth engine project (2011K000925) and a grant from Kangwon Medical Convergence Center of the Ministry of Education, Science and Technology of Korea, and a Korea Research Institute of Bioscience and Biotechnology Research Initiative Program Grant (KGM3100612).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, E., Jeong, M., Singh, R. et al. A chimeric antibody to L1 cell adhesion molecule shows therapeutic effect in an intrahepatic cholangiocarcinoma model. Exp Mol Med 44, 293–302 (2012). https://doi.org/10.3858/emm.2012.44.4.027
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2012.44.4.027
Keywords
This article is cited by
-
An antibody against L1 cell adhesion molecule inhibits cardiotoxicity by regulating persistent DNA damage
Nature Communications (2021)
