
Abstract
Heterozygous variants in BICD cargo adapter 2 (BICD2) cause autosomal dominant spinal muscular atrophy, lower extremity-predominant 2 (SMALED2). The disease is usually characterized by a benign or slowly progressive, congenital or early onset muscle weakness and atrophy that mainly affects the lower extremities, although some affected individuals show involvement of the arms and the shoulder girdle. Here we report unusual extremes of BICD2-related diseases: A severe form of congenital muscular atrophy with arthrogryposis multiplex, respiratory insufficiency and lethality within four months. This was caused by three BICD2 variants, (c.581A>G, p.(Gln194Arg)), (c.1626C>G, p.(Cys542Trp)) and (c.2080C>T, p.(Arg694Cys)), two of which were proven to be de novo. Affected individuals showed reduced fetal movement, weak muscle tone and sparse or no spontaneous activity after birth. Despite assisted ventilation, the condition led to early death. At the other extreme, we identified an asymptomatic woman with a known BICD2 variant (c.2108C>T, p.(Thr703Met)). Radiological examination showed fatty degeneration of selected thigh and calf muscles without clinical consequences. Instead, her son carrying the same variant is affected by a mild childhood onset disease with myopathic and neurogenic features. Mechanisms leading to variable expressivity and onset of BICD2-related disease may include alterations in molecular interactions of BICD2 and suggest the presence of genetic modifiers that may act in a protective fashion to ameliorate or abrogate disease. Our data define an additional severe disease type caused by BICD2 and emphasize a possibly variable etiology of BICD2-opathies with regard to primary muscle and neuronal involvement.
Similar content being viewed by others

Introduction
Heterozygous variants in BICD2 (bicaudal D homolog 2, MIM 609797, NM_015250.3) cause autosomal dominant spinal muscular atrophy with predominant involvement of the lower extremities (SMALED2, MIM 615290).1, 2, 3, 4, 5, 6 BICD2 is a cargo adapter for the intracellular transport of small membranous organelles that interact with the dynein–dynactin motor complex7 and is functionally implicated in retrograde Golgi-ER transport processes.8 Individuals carrying heterozygous BICD2 missense variants usually show benign or slowly progressive muscle weakness and atrophy predominantly of the distal lower limbs with rare involvement of the upper limbs or the shoulder girdle. The majority of described mutations cause a congenital or early childhood onset of disease leading to delayed motor milestones,1, 2, 3, 6 but adult onset has also been reported.3, 5 Most congenital SMALED2 cases present foot deformities and isolated joint contractures, while contractures of the hips and congenital hip dysplasia are described in few individuals.1, 2, 3
Here we describe 3 individuals of independent families with severe arthrogryposis multiplex congenita (AMC), respiratory insufficiency, and early lethality caused by BICD2 (c.2080C>T, p.(Arg694Cys)) and two proven de novo variants (c.581A>G, p.(Gln194Arg)) and (c.1626C>G, p.(Cys542Trp)), respectively. On the other extreme, we describe an asymptomatic women with subclinical findings at high age. Instead, her son carrying the same previously described BICD2 variant (c.2108C>T, p.(Thr703Met))1 presents a mild childhood onset disease with myopathic and neurogenic features. Our data define an additional severe type of disease caused by BICD2, and emphasize a possibly variable etiology of BICD2-opathies with regard to primary muscle and neuronal involvement.
Materials and methods
Whole-exome sequencing
Whole-exome sequencing of family A was performed as trio analysis on genomic DNA isolated from blood. DNA samples were prepared using the NEXTERA Rapid Capture Exome Kit (Illumina, San Diego, CA, USA) and followed by paired-end 150 nt sequencing on the Illumina NextSeq500. Alignment and variant calling was performed as described previously.9 Median coverage of the target region was 95X with 96% of target region covered with at least 10 reads. Data annotation and interpretation were performed using the Cartagenia Bench Lab, NGS module (Cartagenia, Leuven, Belgium). The BICD2 variant was verified by targeted Sanger sequencing. Results of Sanger sequencing were analyzed using the Sequence Pilot software (JSI medical systems GmbH, Ettenheim, Germany).
BICD2 mutational screening in individuals with AMC
Eight affected individuals of healthy parents presenting reduced fetal movement during pregnancy, muscle weakness and atrophy, AMC, and early lethality were selected for mutational screening of BICD2. Genomic DNA of each BICD2 exon was amplified by PCR and sequenced by routine Sanger sequencing as previously described.1 Fragments were sequenced on an ABI 3500 Series Genetic Analyzer and data were aligned to the BICD2 transcript ENST00000356884 (NM_015250.3) and analyzed using the Sequence Pilot software (JSI medical systems GmbH, Ettenheim, Germany). Identified variants in BICD2 were validated by bidirectional sequencing and segregation analysis was performed if parental DNA was available.
Variant assessment
Identified variations were analyzed by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and Mutation Taster (http://www.mutationtaster.org/) to predict the pathogenicity of the respective variant. The variant frequency in the healthy control population was evaluated using ExAC (http://exac.broadinstitute.org/) and gnomAD (http://gnomad.broadinstitute.org/), respectively. Amino acid conservation of the altered BICD2 protein regions was analyzed using a multispecies alignment comparing human, monkeys, smaller mammals, chicken, fish, frog, fly and worm. Protein sequences were obtained from Uniprot (http://www.uniprot.org/) and alignments were generated using the Lasergene MegAlign Software (DNASTAR, Madison, WI, USA). Variants identified in this study were uploaded to the Leiden Open Variation Database (LOVD) under the individual identifiers 00101385 to 00101388 and are available under https://databases.lovd.nl/shared/variants/BICD2/unique.
Gene panel analysis
Individual D-II.1 was tested for mutations in multiple disease-causing genes. The AR gene for spinobulbar SMA and deletions of the SMN1 gene for 5q-associated SMA were analyzed at the University Hospital of Aachen, Germany. Pathogenic variants in the RYR1 gene (central core disease and neuromuscular disease) were excluded at the University of Würzburg. Moreover, gene panel analyses have been carried out for 27 SMA-associated genes (panel NMD01), 73 genes associated with congenital and distal myopathies (panel NMD01), and 31 muscular dystrophy-associated genes (panel NMD05) at the Center for Genomics and Transcriptomics (CeGaT, Tübingen, Germany).
RNA expression analysis
RNA of individual D-I.2 was isolated from a PAXgene blood sample using the PAXgene Blood RNA Kit version 2 (PreAnalytiX, Hombrechtikon, Switzerland). 300 ng RNA were reversely transcribed using the QuantiTect Reverse Transcription kit (Qiagen, Hilden, Germany). The BICD2 region containing the (c.2108C>T, p.(Thr703Met)) variant was amplified from cDNA using the forward oligonucleotide 5′-CGTGGACAAGGACAAGGAAGCG-3′ and the reverse oligonucleotide 5′-GCTCTCACAAGTGTCCTGCTGATG-3′. The PCR product was sequenced by Sanger sequencing to qualitatively detect biallelic expression of BICD2 in the asymptomatic individual D-I.2.
Muscle biopsy and electron microscopy
Muscle biopsy of individual A-II.1 was taken from the right vastus lateralis muscle at the age of 1 month and directly frozen at −80 °C. For conventional histological analysis, a part of the biopsy was cryo-sectioned using Leica CM3050 cryomicrotome (Mannheim, Germany). 5-7 μm sections were fixed in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) for 10 minutes and standard clinical H&E, Trichrome and ATPase staining was performed. For electron microscopy, samples were fixed in 100 mM phosphate buffer (PB) containing 4% PFA, 1% glutaraldehyde and 15% picric acid, pH 7.4, at 4 °C overnight. Samples were cut into 50 μm longitudinal sections using a VT 1000 S Leica vibratome (Mannheim, Germany), before rinsing twice in PBS. Following treatment with 0.5% OsO4 for 45 min and several washing steps in 100 mM PBS, samples were counterstained with uranyl acetate, dehydrated via ethanol series, and embedded in Durcupan ACM epoxy resin (Fluka, Switzerland). Ultrathin sections were prepared from resin blocks using a Leica Ultracut S (Mannheim, Germany) and adsorbed to glow-discharged formvar-carbon-coated copper single slot grids. Electron micrographs were recorded using a ZEISS LEO 910 electron microscope; images were taken with a TRS sharpeye CCD camera (Troendle, Moorenwies, Germany).
Muscle magnetic resonance imaging
Magnetic resonance imaging (MRI) is an objective and non-invasive measure, which can be used to identify distinct patterns of muscle involvement in hereditary neuromuscular diseases. MRI in individual D-I.2 was performed on a 1.5 Tesla MR scanner generating axial and coronal T1-weighted images to detect fat replacement and STIR (Short-Tau-Inversion-Recovery) sequences to detect edematous muscle alterations of the lower limbs. Fatty tissue replacement appears white on T1-weighted images.
Ethical standards
Written informed consent for study participation, analyses of proband material, photographs, and publication of anonymized data was obtained from affected individuals and parents. The NeurOmics study was approved by the ethical committee of the University of Cologne.
Results
Variants in BICD2 are predominantly associated with the SMALED2 phenotype,1, 2, 3, 4, 5, 6 and contractures or milder forms of congenital arthrogryposis have been described in affected individuals. Here we describe three cases two of them with proven de novo variants in BICD2 causing a severe condition that is different from SMALED2 including fetal hypokinesia, AMC, hypotonia, respiratory failure and early lethality at one extreme, and one neurologically unaffected individual with subclinical findings on MRI at high age carrying a known BICD2 variant at the other extreme.
Individual A-II.1
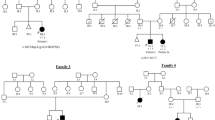
We identified a de novo BICD2 variant (c.581A>G, p.(Gln194Arg)) by whole-exome sequencing in a Norwegian affected individual (A-II.1) with severe disease including profound hypotonia, AMC and respiratory failure with lethal outcome (Figures 1a and 2a–c; Table 1). Larger chromosomal aberrations were excluded by aCGH (array comparative genomic hybridization) analysis, and Sanger sequencing of both parents confirmed the de novo nature of the BICD2 variant (Figure 1b). The identified variant was absent from the ExAC or gnomAD database. The glutamine residue at position 194 is highly conserved among species (Figure 1c) and its substitution to arginine was rated disease causing by Mutation Taster, probably damaging (0.969) by PolyPhen-2 and tolerated (0.41) by SIFT. Individual A-II.1 (Figures 2a–c) was a girl of healthy unrelated parents born at GW40+1 with Apgar scores of 4/5/7. The girl presented reduced muscle tone, reduced spontaneous movement and weak respiration. Hourly nasopharyngeal suction for mucus mobilization and persistent CPAP was required to keep peripheral O2 saturation above 90%. The girl showed severe AMC with bilateral hip dislocation and multiple joint deformities including overriding fingers at both hands (Figures 2a and b), right-sided pes equinovarus, varus deformity of the left ankle and scoliotic appearance, but no skeletal spine deformity. There were no facial abnormalities, the girl appeared alert and focused when awake. The heart was structurally normal except for a haemodynamically significant patent ductus arteriosus, which was treated with diuretics and ACE inhibitors from 3 months of age. Cerebral magnetic resonance imaging (MRI) showed a moderate thinning of the corpus callosum and two minor lesions adjacent to the posterior part of both second ventricles of unknown significance. Electroencephalography (EEG) did not reveal seizure activity, even though it was clinically suspected on several occasions. The girl could not eat unassisted, but tolerated mother’s milk through a nasogastric tube. The weight increase was slow. The girl deceased at 4 months of age due to acute respiratory failure. Muscle biopsy obtained at one month of age showed severe myopathic features with massive atrophy, many hypotrophic and necrotic fibers, and increased fibrous and fatty connective tissue, the latter with macrophagic infiltrations (Figure 2d). A perimysial and endomysial fibrosis as well as an increased number of muscle fibers with central nuclei and nuclear clumps was observed. ATPase staining revealed many Type 2 muscle fibers showing low oxidative activity (Figure 2e).

Families with heterozygous variants in BICD2. (a) Families and identified variants in BICD2. Individuals with gray outline in the pedigree were not included into segregation analysis, as no DNA was available. (b) Sanger sequencing results showing the respective locus of the identified BICD2 variant in the affected individual and in the parents if available. (c) Interspecies alignment of the BICD2 protein region harboring the respective mutation. The upper panel indicates the conservation score in color code. Black arrowhead and blue shading mark the mutated residue. Aligned species include: Homo sapiens, Pan troglodytes (chimpanzee), Macaca mulatta (Rhesus macaque), Felis catus (cat), Rattus norvegicus (rat), Mus musculus (mouse), Gallus gallus (chicken), Takifugu rubripes (pufferfish), Danio rerio (zebrafish), Drosophila melanogaster (fruit fly), Caenorhabditis elegans (worm) and Xenopus tropicalis (frog). Afg, global allele frequency in the gnomAD database. The number in parentheses indicates the absolute number of variant alleles in the gnomAD database. W, wild-type allele; M, mutant allele; N/A, not available.

Affected individual A-II.1 carrying the BICD2 (c.581A>G, p.(Gln194Arg)) variant. (a–c) Photographs of individual A-II.1. (d–g) Histological analysis of muscle biopsy of affected individual A-II.1 (d, e) and an unaffected control (f, g). Biopsy sections were stained with hematoxylin and eosin and for ATPase (d, f). (h–o) Electron microscopy analysis of muscle biopsy material obtained from affected individual A-II.1 at one month of age showed an advanced stage of myofibrillar breakdown (h), with lesions and a massive presence of lysosomal vesicles, accompanied by enlarged polymorphic mitochondria and abundant autophagic vacuoles (j). Most of the necrotic myocytes contained dissociated myofibrils accompanied by enlarged polymorphic mitochondria, only a few sarcomeres had preserved Z-discs. The contractile apparatus showed only a minor structural integrity, the sarcolemma appeared with perforations and leaks (k, inlet arrows). Dispersed dictyosomes (ER, Golgi apparatus) were observed (I, inlet) in the nuclear vicinity, which are usually only rarely visible in control muscle tissue (m). The extracellular space showed a massive deposit of collagen fibrosis. Large numbers of various vesicles were present near the sarcolemma, the overall observed subsarcolemmal vesicle localization und membrane fusion represented a massive exocytosis and a thick (20–50 μm) extracellular electron dense layer deposition on the sarcolemma, which was not observed in normal human control muscle tissue (o). Age-matched control samples of an unaffected individual (l, m) and of an individual affected by (neurogenic) spinal muscular atrophy (n, o) appeared normal without any obvious pathological findings in myofibrillar integrity. MC, myocyte; SL, sarcolemma; ER, endoplasmic reticulum; GA, Golgi apparatus; NC, nucleus; MF, myofibril; scale bar is 40 μm.
Electron microscopical analysis of muscle biopsy indicated an advanced stage of myofibrillar breakdown (Figure 2h) with lesions, a massive presence of lysosomal vesicles and abundant autophagic vacuoles (Figure 2j). The contractile apparatus showed only a minor structural integrity, only a few sarcomeres with preserved Z-discs were observed accompanied by enlarged polymorphic mitochondria, the sarcolemma appeared with perforations and leaks (Figure 2k). The Golgi apparatus appeared widely enlarged compared with unaffected controls (Figures 2i and m). This is in line with previous findings obtained for other BICD2 mutations in muscle tissue of affected individuals,10 in HeLa cells harboring mutated BICD2 constructs, and in primary fibroblasts derived from affected individuals.1 Control samples of an unaffected individual (Figures 2l and m) and of an individual with neurogenic neuromuscular disease (SMA, Figures 2n and o) appeared with normal muscle architecture and without any pathological findings. This suggests a myopathic role of BICD2 (c.581A>G, p.(Gln194Arg)) perturbing structure and function of the contractile apparatus of the muscle. This feature may be the primary cause of the disease or may act independently of possible neuropathologic features caused by BICD2 variants present in SMALED2.
Based on the extreme severity of the BICD2-related phenotype in this affected individual, we screened eight individuals with severe spinal muscular atrophy, severe arthrogryposis, respiratory insufficiency and early lethality for possible pathogenic variants in BICD2. All 8 affected individuals had healthy parents. Homozygous deletion of SMN1 (survival of motor neuron 1) had previously been excluded as the cause of disease by MLPA (multiplex ligation-dependent probe amplification). All BICD2 exons were analyzed by Sanger sequencing from genomic DNA isolated from peripheral blood.
Individual B-II.1
We identified the heterozygous BICD2 missense variant (c.1626C>G, p.(Cys542Trp)) in the affected individual B-II.1 (Figures 1a and b; Table 1). The variant was not listed in the ExAC or gnomAD database and the amino acid substitution cysteine to tryptophan at position 542 lies within a protein region that is highly conserved among species (Figure 1c). Accordingly, the substitution was classified as disease causing, damaging (score=0), and probably damaging (score=1) by Mutation Taster, SIFT, and Poly-Phen2, respectively. Segregation analysis indicated that this variant arose de novo, as it was not present in either parent (Figure 1b). Ultrasound of individual B-II.1 during pregnancy showed contractures, enlarged head, and macroglossia. Polyhydramnios was diagnosed and fetal movements were normal at first but decreased gradually until birth by cesarean section at 32+2 weeks. After birth, the affected individual presented with reduced movements, a heart rate of 50 bpm, and was stabilized under assisted ventilation (Apgar 6/7/8). AMC presented as contractures of arms and shoulders, clinodactyly and adducted thumbs, hip dislocation with contractures, and luxation of the ankle joints with contractures. Respiration was weak with permanent need for assisted ventilation, cardiac function was normal. Spontaneous activity and reflex-responses to pain-stimuli were absent from all limbs. Sensitivity was obviously intact, as pain stimulation resulted in hyperventilation. Deep tendon reflexes were absent. During the first week of life, spontaneous activity remained absent. Despite difficult interpretability, electromyography pointed to a neurogenic generalized muscular atrophy. A muscle biopsy showed hypotrophic and atrophic fibers. Based on a poor prognosis, individual B-II.1 deceased upon suspending intensive care measures after 15 days of life.
Affected individual C-II.1
Sanger sequencing of individual C-II.1 revealed the heterozygous BICD2 missense variant (c.2080C>T, p.(Arg694Cys)) (Figures 1a and b; Table 1). As parental DNA was not available, segregation analysis was not performed. This causative variant has recently been reported as recurrent de novo with very severe congenital disease in two independent families,11 and given that both parents of individual C-II.1 were unaffected, we assume that the BICD2 variant (c.2080C>T, p.(Arg694Cys)) arose de novo in family C as well. The variant was not listed in the ExAC or gnomAD database and arginine at position 694 is conserved in all assayed species (Figure 1c). In concordance with the clinical reports of Ravenscroft et al.11 the newborn (GW36) presented with AMC, clenched hands, micrognathia, and respiratory insufficiency that required permanent assisted ventilation. Individual C-II.1 showed strongly diminished spontaneous movement of the upper limbs, while spontaneous activity was absent from the lower limbs. Head control was diminished and pectus excavatum was present. Electromyography showed a clear neurogenic pathology with signs of denervation without any pathological spontaneous activity. A muscle biopsy was inconclusive, as it contained primarily fat and connective tissue. Brain MRI was not performed for individual C-II.1, so we cannot comment on the association with bilateral symmetric perisylvian polymicrogyria, which has been reported for BICD2 (c.2080C>T, p.(Arg694Cys)).11
The BICD2 (c.2108C>T, p.(Thr703Met)) variant causes myopathic changes in an asymptomatic individual D-I.2
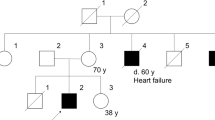
We recently reported about individual D-II.1 in the work of Unger and colleagues.10 Individual D-II.1 is of German descent and carrier of the disease-causing BICD2 variant (c.2108C>T, p.(Thr703Met)). He presented childhood onset disease with primarily myopathic alterations, but only minor neurogenic changes, indicating a combined involvement of muscle and neuronal systems.10 In contrast to individuals that carry the BICD2 (c.2108C>T, p.(Thr703Met)) variant and are affected by SMALED2,1, 12 affected individual D-II.1 showed no contractures, later onset, and milder course of disease. Proximal lower limb muscle strength and the patellar reflex were preserved, while the ankle jerk reflex was absent and distal muscles were atrophied. Lower limb muscle MRI of individual D-II.1 showed fatty replacement in selected thigh and calf muscles (not shown) and the CK level was increased to 581U/l. The affected individual D-II.1 could walk on toes, but not on heels. There was no general impairment in accomplishment of every day needs.
For this family, we now report dominant inheritance of the BICD2 (c.2108C>T, p.(Thr703Met)) variant from the neurologically unaffected mother D-I.2 (Figures 1a and b), who was clinically asymptomatic, but presented myopathic features on MRI (Figure 3a). To our knowledge, this has yet not been reported for carriers of established disease-causing mutations in BICD2. This also raised doubt on the BICD2 (c.2108C>T, p.(Thr703Met)) variant being the real reason for the son’s (D-II.1) disease. In a search for possible other causes of disease, multiple genetic testing approaches for known disease-causing genes have been performed on the affected son D-II.1: No disease-causing variants were detected in genes causing 5q-associated SMA (SMN1), spinobulbar SMA (AR) and central core disease/neuromuscular disease (RYR1). Moreover, gene panel analyses have been performed for 27 SMA-associated genes, 73 genes associated with congenital and distal myopathies, and 31 muscular dystrophy-associated genes. Though whole-exome or genome sequencing would be required to fully rule out other causative genes for disease individual D-II.1, the negative results of these investigations in addition to previous functional studies of this variant1 support the idea that the identified BICD2 variant (c.2108C>T, p.(Thr703Met)) is indeed causative for the disease. The amino acid position Thr703 is highly conserved among species and former functional studies support pathogenicity,1 but in contrast to all other BICD2 variants discussed here, it is listed in the gnomAD database with a global frequency of 7.1 × 10-6 (that is, two chromosomes). This might support the rare possibility that young, seemingly unaffected individuals may be carriers of this disease-causing variant. The mother (D-I.2), aged 71, did not seek medical attention for muscular problems, until the genetic testing confirmed the same BICD2 variant as in her affected son. She could walk without any impairment and also walking on heels as well as standing and jumping on one leg was possible without problems. She had no sensory impairment and deep tendon reflexes were fully preserved. Walking on toes and climbing stairs was slightly impaired. CK levels of 170 U/l and 47 U/l, respectively, were within normal range. Her neurological examination showed a slight symmetrical distal muscle weakness for plantar flexion (MRC grade 4+/5) without any other abnormalities. We have now performed MRI studies of individual D-I.2 (Figure 3a), which revealed mild alterations with a symmetrical fatty degeneration of the rectus femoris and satorius thigh muscles, without signs of muscle edema. At the calf level, gastrocnemii muscles showed advanced fatty degeneration, but there were only mild fatty changes of the extensor digitorum muscle. An age-matched control individual did not show this specific pattern of fatty degeneration, indicating the muscle degeneration is not age-related (Figure 3a). These results indicate that – even though clearly milder than in her son – also the mother showed pathological findings that did not result in clinically apparent muscular weakness, however.

Myopathic changes caused by the BICD2 variant (c.2108C>T, p.(Thr703Met)). (a) MRI examination (1,5T; transversal T1-weighted and STIR (Short-Tau-Inversion-Recovery)) of the mildly affected individual D-I.2 carrying the disease-causing BICD2 mutation p.(Thr703Met) and an age-matched control individual. Studies showed a symmetrical fatty degeneration (white areas) of the rectus femoris (arrow heads) and sartorius (asterisks) muscles of the thigh, without signs of muscle edema. At the calf level, gastrocnemii muscles (hashes) showed advanced fatty degeneration with only mild fatty changes of the extensor digitorum muscles. Changes were not present in the control individual. (b) RNA expression analysis of BICD2 transcript in whole blood. The mutant BICD2 transcript carrying the (c.2108C>T, p.(Thr703Met)) variant is expressed in the asymptomatic mother. Afg, global allele frequency.
To qualitatively confirm biallelic expression of BICD2 in the asymptomatic mother D-I.2, we reversely transcribed whole blood RNA and sequenced the BICD2 cDNA region comprising the variant. Sanger sequencing results indicated the expression of both the wild-type and mutant allele in the asymptomatic mother (Figure 3b). These results suggest that the mutant BICD2 allele is expressed in the asymptomatic mother and thus, her mild disease likely cannot be attributed to diminished expression of the mutant transcript.
Discussion
Here we expand the phenotypical spectrum of BICD2-opathies into both directions of disease severity: First, we identified the de novo missense variants (Figure 4) (c.581A>G, p.(Gln194Arg)), (c.1626C>G, p.(Cys542Trp)) and most likely (c.2080C>T, p.(Arg694Cys)) in individuals with severe congenital neuromuscular disease and arthrogryposis multiplex, respiratory insufficiency and early lethality – a phenotype of drastically increased severity as compared with the typical SMALED2 presentation. Second, we demonstrate subclinical changes in an asymptomatic carrier of the known BICD2 disease-causing mutation (c.2108C>T, p.(Thr703Met)) at high age.

Protein structure and disease-causing BICD2 mutations. The domain structure of the BICD2 protein comprises three coiled-coil (CC) domains. Numbers above the domains indicate amino acid positions. The protein domain conservation among species is given in color code. The upper panel summarizes known interaction partners of the respective domains.19 The lower panel represents disease-causing variants in BICD2 on cDNA and protein level.1, 2, 3, 4, 5, 6, 11, 18 Boxed variants are subject of this work. CC, coiled-coil domain; aa, amino acid; HC, heavy chain. Asterisks indicate disease associations made in this study.
Within the peripheral nervous system, degeneration of anterior horn cells, axonal or demyelinating neuropathies, and blockage of the neuromuscular junction may cause arthrogryposis, which is associated with reduced fetal movement.13 In line with this, arthrogryposis is observed for example in congenital myasthenic syndromes,14 in severe cases of autosomal recessive spinal muscular atrophy (SMA),15 and in X-linked SMA.16 Isolated congenital contractures can be as common as one in 500 live births17 and isolated contractures are indeed a reported feature in individuals with SMALED2 as well.1, 2, 3 Arthrogryposis may be of neurogenic or myopathic origin.13 The severe cases described here, present as AMC and unlike in SMALED2, the affected muscle groups are not primarily restricted to the distal lower extremities. Even though, particularly in individual C-II.1, the lower extremities were still more severely affected than the upper limbs, there is a rather general muscular weakness and atrophy affecting the whole body of most severely affected infants. Notably, individual A-II.1 showed an advanced stage of myofibrillar breakdown in the vastus lateralis muscle with only few Z-discs preserved. This indicated a pathomechanism that is different from a neurogenic muscular atrophy suggesting a strong myopathic involvement for the BICD2 mutation p.(Gln194Arg). In contrast, EMG data of affected individuals B-II.1 and C-II.1 carrying the BICD2 mutations p.(Cys542Trp) and p.(Arg694Cys), respectively, suggested a neurogenic muscular atrophy with signs of denervation. There was no EMG performed on individual A-II.1, and no conclusive muscle biopsy result or electron micrograph is available of individuals B-II.1 and C-II.1 (Table 1). Thus, we can neither exclude a myopathic, nor a neuropathic involvement. A dual mode of pathology affecting nerve and muscle independently, as well as prenatal early onset motor neuron disease perturbing muscle development, are possible scenarios that may apply to each BICD2 variant causing severe disease.
Recently, Ravenscroft and colleagues have reported a recurrent de novo mutation p.(Arg694Cys) in BICD2 in two individuals with congenital arthrogryposis multiplex with congenital fractures, generalized hypotonia, respiratory insufficiency, and bilateral perisylvian polymicrogyria.11 Respiratory insufficiency required permanent CPAP or tracheotomy and resulted in fatal outcome after 45 days in one of the two affected individuals. Interestingly, this is the same variant as the one found in individual C-II.1, suggesting that this variant is strongly related to a severe disease manifestation. Another de novo BICD2 mutation p.(Leu683Arg) has been found to cause SMALED2 plus cerebellar hypoplasia.18
The understanding of the BICD2-related pathomechanism is very limited at present and the localization of the disease-causing mutations along the protein does not seem to be in correlation with the phenotypical presentation and severity of disease. The BICD2 mutation p.(Gln194Arg) identified in the Norwegian individual localizes to the N-terminal coiled-coil 1 (CC1) domain. Two other known disease-causing mutations, p.(Asn188Thr) and p.(Ile189Phe), are located in the immediate vicinity. Though there are few cases described with these two mutations,1, 3, 4 neither one causes a disease of comparable severity. In particular, the p.(Asn188Thr) mutation has been reported to cause a relatively mild form of SMALED2.1 Similarly, the other mutations p.(Cys542Trp) and p.(Arg694Cys) causing severe BICD2-opathy with early lethality have known disease-causing mutations with a milder disease phenotype in their nearby regions (Figure 4).19 This suggests that specific amino acid residues of BICD2 must be pivotal and might have particular impact on conformation, molecular interactions with other proteins, or other protein regions of BICD2 itself. Indeed, Hoogenraad and colleagues demonstrated intramolecular interactions between the CC1 and CC3 domain of BICD2.7 Such intramolecular interactions could be a regulatory feature for the interaction of BICD2 with other partners like the dynein–dynactin complex or the RAB6a GTPase that have their interaction sites in the CC1 and CC3 domains of BICD2, respectively.8, 19, 20 BICD2 is considered a cargo-linking protein, which couples a cargo bound to the C-terminal region (RAB6a vesicles) to the dynein–dynactin motor complex associated with the N-terminal region of BICD2.7, 21 Disturbed intracellular transport processes hold the potential to cause failure of organization, growth and maintenance in neurons and muscle cells and are thus, one possible explanation for BICD2-related disease.
Interestingly, the cortical malformations previously described in association with BICD2 mutations, overlap phenotypically with neurological disorders due to variants in the DYNC1H1 gene (MIM 600112), which encodes a large crucial subunit of the cytoplasmic dynein complex.22 Similarly to BICD2, mutations in DYNC1H1 have been described in cases with phenotypical extremes. Mutations in DYNC1H1 are described as causative for malformations of cortical development (MCD MIM 614563) and experimental evidence supports that these mutations decrease dramatically the dynein motility, and likely impair neuronal proliferation or migration during development of the cerebral cortex.23, 24 In contrast, mutations in DYNC1H1 may also cause SMALED1 (MIM 158600), a much milder phenotype that affects lower extremities predominantly without any cortical malformation.24, 25
Impaired cellular transport processes may be associated to vastly enlarged or fragmented Golgi structures in affected muscle tissue and primary fibroblasts. Golgi fragmentation was previously reported for mutations causing mild BICD2-opathies including SMALED2 and chronic myopathy.1, 10 In the current study, we present a similar impairment of Golgi integrity in a severely affected patient (Figure 2i). This suggests that – despite being clinically different – both diseases may be based on the same molecular pathomechanism. Detailed molecular interaction studies will be needed to delineate alterations of inter- and intramolecular interactions and their possible effects on intracellular transport processes in the presence of mutations that cause severe disease.
On the other side of the phenotypical spectrum, we report an asymptomatic carrier of the known disease-causing BICD2 mutation p.(Thr703Met) with myopathic features in MRI, which to our knowledge has not been reported before. However, variable severity of disease is a well-known feature of many known disease-causing variants (reviewed by Martinez-Carrera and Wirth).19 To our knowledge, only three individuals of 2 families have been reported with the p.(Thr703Met) mutation.1, 10 Two affected individuals (father and son) of a Canadian family1 had SMALED2 that was more severe than the BICD2-opathy with myopathic involvement in individual D-II.1.10 The mother D-I.2 was even asymptomatic without neurogenic pathology, but showed fatty replacement in the lower extremities at high age. Even though the BICD2 mutation p.(Thr703Met) has been reported disease-causing before, we cannot exclude other genetic or environmental effects in family D that may act synergistically.26 Furthermore, the BICD2 variant (c.2108C>T, p.(Thr703Met)) is rare, but not absent from the gnomAD database, which suggests that there are seemingly healthy or asymptomatic carriers of this disease-causing variant. Variable expressivity of disease and possibly incomplete penetrance at younger age suggest the presence of genetic modifiers that may act in a protective fashion to ameliorate or abrogate disease. Indeed, we recently described the power of two genetic modifiers for autosomal recessive spinal muscular atrophy.27, 28 Studies in SMA mouse models suggested that genetic modifiers might turn severe cases into mild cases or turn mild cases asymptomatic. The presence of variable disease severity in different families and additionally the possibility of incomplete penetrance suggest that multiple modifying elements exist and may act synergistically. However, the identification of modifying variants in singletons is virtually impossible and a functional proof extremely laborious. In contrast, the three BICD2 variants causing severe congenital muscular atrophy with arthrogryposis are absent from the gnomAD database and arose de novo in each family. This further supports the idea that BICD2 mutations causing severe muscular atrophy, AMC and respiratory distress have a strong and unprecedented impact on the protein function and thus, appear largely incompatible with life.
References
Neveling K, Martinez-Carrera LA, Holker I et al: Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 2013; 96: 946–954.
Peeters K, Litvinenko I, Asselbergh B et al: Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am J Hum Genet 2013; 92: 955–964.
Oates EC, Rossor AM, Hafezparast M et al: Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 2013; 92: 965–973.
Rossor AM, Oates EC, Salter HK et al: Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain 2015; 138: 293–310.
Synofzik M, Martinez-Carrera LA, Lindig T, Schols L, Wirth B : Dominant spinal muscular atrophy due to BICD2: a novel mutation refines the phenotype. J Neurol Neurosurg Psychiatry 2014; 85: 590–592.
Rudnik-Schoneborn S, Deden F, Eggermann K et al: Autosomal dominant spinal muscular atrophy with lower extremity predominance: a recognizable phenotype of BICD2 mutations. Muscle Nerve 2016; 54: 496–500.
Hoogenraad CC, Akhmanova A, Howell SA et al: Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J 2001; 20: 4041–4054.
Matanis T, Akhmanova A, Wulf P et al: Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat Cell Biol 2002; 4: 986–992.
Bredrup C, Johansson S, Bindoff LA et al: High myopia-excavated optic disc anomaly associated with a frameshift mutation in the MYC-binding protein 2 gene (MYCBP2). Am J Ophthalmol 2015; 159: 973–979, e972.
Unger A, Dekomien G, Guttsches A et al: Expanding the phenotype of BICD2 mutations toward skeletal muscle involvement. Neurology 2016; 87: 2235–2243.
Ravenscroft G, Di Donato N, Hahn G et al: Recurrent de novo BICD2 mutation associated with arthrogryposis multiplex congenita and bilateral perisylvian polymicrogyria. Neuromuscul Disord 2016; 26: 744–748.
Adams C, Suchowersky O, Lowry RB : Congenital autosomal dominant distal spinal muscular atrophy. Neuromuscul Disord 1998; 8: 405–408.
Bamshad M, Van Heest AE : Pleasure D: Arthrogryposis: a review and update. J Bone Joint Surg Am 2009; 91: 40–46.
Abicht A, Muller JS, Lochmuller H Congenital Myasthenic Syndromes. In: Pagon RA, Adam MP, Ardinger HH et al(eds): GeneReviews(R) 1993.
Burglen L, Amiel J, Viollet L et al: Survival motor neuron gene deletion in the arthrogryposis multiplex congenita-spinal muscular atrophy association. J Clin Invest 1996; 98: 1130–1132.
Ramser J, Ahearn ME, Lenski C et al: Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet 2008; 82: 188–193.
Hall JG : Arthrogryposis multiplex congenita: etiology, genetics, classification, diagnostic approach, and general aspects. J Pediatr Orthop B 1997; 6: 159–166.
Fiorillo C, Moro F, Brisca G et al: Beyond spinal muscular atrophy with lower extremity dominance: cerebellar hypoplasia associated with a novel mutation in BICD2. Eur J Neurol 2016; 23: e19–e21.
Martinez-Carrera LA, Wirth B : Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein. Front Neurosci 2015; 9: 401.
Splinter D, Razafsky DS, Schlager MA et al: BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol Biol Cell 2012; 23: 4226–4241.
Terenzio M, Schiavo G : The more, the better: the BICD family gets bigger. EMBO J 2010; 29: 1625–1626.
Poirier K, Lebrun N, Broix L et al: Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 2013; 45: 639–647.
Schiavo G, Greensmith L, Hafezparast M, Fisher EM : Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci 2013; 36: 641–651.
Hoang HT, Schlager MA, Carter AP, Bullock SL : DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes. Proc Natl Acad Sci USA 2017; 9: e1597–e1606.
Harms MB, Ori-McKenney KM, Scoto M et al: Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 2012; 78: 1714–1720.
Kousi M, Katsanis N : Genetic modifiers and oligogenic inheritance. Cold Spring Harb Perspect Med 2015; 5: a017145.
Hosseinibarkooie S, Peters M, Torres-Benito L et al: The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet 2016; 99: 647–665.
Riessland M, Kaczmarek A, Schneider S et al: Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet 2017; 100: 297–315.
Acknowledgements
We are grateful to the families, who supported this study. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 2012-305121 “Integrated European –omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS)”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Storbeck, M., Horsberg Eriksen, B., Unger, A. et al. Phenotypic extremes of BICD2-opathies: from lethal, congenital muscular atrophy with arthrogryposis to asymptomatic with subclinical features. Eur J Hum Genet 25, 1040–1048 (2017). https://doi.org/10.1038/ejhg.2017.98
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.98
This article is cited by
-
A homozygous loss-of-function variant in BICD2 is associated with lissencephaly and cerebellar hypoplasia
Journal of Human Genetics (2022)
-
Loss of BICD2 in muscle drives motor neuron loss in a developmental form of spinal muscular atrophy
Acta Neuropathologica Communications (2020)
-
Impairment in dynein-mediated nuclear translocation by BICD2 C-terminal truncation leads to neuronal migration defect and human brain malformation
Acta Neuropathologica Communications (2020)
-
Dynein activating adaptor BICD2 controls radial migration of upper-layer cortical neurons in vivo
Acta Neuropathologica Communications (2019)
