
Abstract
CHES (cerebellar hypoplasia with endosteal sclerosis) syndrome (OMIM#213002) associates hypomyelination, cerebellar atrophy, hypogonadism and hypodontia. So far, only five patients have been described. The condition is of neonatal onset. Patients have severe psychomotor delay and moderate to severe intellectual disability. Inheritance is assumed to be autosomal recessive due to recurrence in sibs, consanguinity of parents and absence of vertical transmission. CHES syndrome is reminiscent of 4H-leukodystrophy, a recessive-inherited affection due to variations in genes encoding subunits of the RNA polymerase III (POLR3A–POLR3B–POLR1C). POLR3B variants have been identified in one CHES patient. Here we report on a novel CHES patient, carrying compound heterozygous variations in POLR3B. This report confirms affiliation of CHES to POLR3-related disorders and suggests that CHES syndrome represents a severe form of 4H-leukodystrophy.
Similar content being viewed by others

Introduction
Cerebellar hypoplasia with endosteal sclerosis (CHES) was first reported by Stoll et al1 in a single patient as a possible hitherto unrecognized entity. Subsequently, two affected sibs born to consanguineous parents and an unrelated third patient were reported, suggesting autosomal recessive inheritance.2 A fifth patient has been reported with a long-term follow-up.3 Neurological features are characterized by moderate to severe developmental delay, progressive ataxia and global cerebellar atrophy. Bone changes comprise endosteal and metaphyseal abnormalities, inducing narrowness of the medullary cavity of long bones. Sclerosis is variable and remains stable or decreases slightly with age. Eruption pattern of primary teeth is unusual and patients may have missing teeth. Myopia and hearing loss can occur (Table 1). Here we report on a sixth patient with CHES, provide further information on the phenotype, and add evidence that the disorder is caused by POLR3B variants, widening the POLR3-related phenotypic spectrum.4, 5
Case report
The patient was born from healthy unrelated parents after an uneventful pregnancy. Family history was unremarkable. At birth the patient was markedly hypotonic. Length, weight and skull circumference were normal. Sitting was achieved at 10 months. Independent walking was never acquired. In the first year, tremor and dysmetria were noticed. Thereafter ataxia worsened rapidly, leading to severe intention tremor, severe dysmetria, dysarthria and gait ataxia. Language development was also severely impaired. Although no formal testing was performed, his examination was consistent with severe cognitive impairment. The patient had five teeth at birth. Subsequently his primary molar erupted earlier than his incisors, and eventually he was missing four superior molars and two inferior premolars. At 15 years he showed growth retardation (height 138 cm (−2 SD), weight 31 kg (−2 SD) but no microcephaly (OFC 52 cm (−1 SD)). He remained hypotonic and he developed pyramidal signs and dystonia. He was wheelchair-bound, independent gait not being possible. There was no facial dysmorphism. He was prepubertal and had under-developed external genitalia.
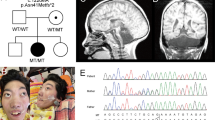
A brain MRI performed at 15 years showed diffuse hypomyelination, severe cerebellar atrophy and T2-weighted images reminiscent of 4H-leukodystrophy (Figure 1). Skeletal X-rays at 2 years old demonstrated vertebral, pelvic and femoral endosteal sclerosis, and left hip dislocation. At 15 years the endosteal sclerosis had improved (Figure 1). Ophthalmological examination revealed high myopia (−9 Dioptries) and optic atrophy. Endocrine tests showed hypogonadotropic hypogonadism.

Brain MRI performed at 15 years old showing hypopmyelination (a–d) and severe cerebellar atrophy (b, c, e, f, h, i). T2-weighted images show hypointensity of globi pallidi, ventrolateral thalami, as well as optic radiations and posterior limb of the internal capsule (g, white arrows). At the pons level, T2 hypointensity of the medial lemniscus was observed (e, white arrow). Cerebellar white matter showed mild T2 hyperintensity. Thinning of the corpus callosum was also observed (h, i). Skeletal X-rays performed at 15 years old show endosteal sclerosis involving pelvis, vertebrae and long bones, causing reduction of the medullary cavity (k–m). Comparison of pelvis X-rays performed at 2 years (l) and 15 years old (m) showing that endosteal sclerosis slightly decreased with age.
Genetic testing
Because of overlapping phenotypes between CHES and 4H-leukodystrophy (OMIM #614381), genetic testing of POLR3A, POLR3B was performed in the patient reported by Ozgen et al,3 allowing the identification of compound heterozygous variations in POLR3B NM_018082.5(POLR3B):c.[303+1G>A];[1568T>A] p.[(?)];[(Val523Glu)] (GRCh37 Chr12:g.[106763129G>A];[106826199T>A]).4 In the present patient, Sanger sequencing identified compound heterozygous variations in POLR3B NM_018082.5(POLR3B):c.[1568T>A];[2774C>A] p.[(Val523Glu)];[(Pro925Gln)] (GRCh37 Chr12:g.[106826199T>A];[106889893C>A]) (Figure 2). Missense variant c.2774C>A p.(Pro925Gln) has never been reported and is not present in the ExAC database (http://exac.broadinstitute.org/). In silico predictions classified the variation as 'damaging'. It induces the substitution of a highly conserved-across-species Pro residue by a Gln residue (Figure 2). Structure of human RNA polymerase III (POL III) and yeast RNA polymerase II (POL II) is highly homologous. Then, based on the extrapolation of POLR3B (RPC2 subunit of POL III) from the yeast Pol II structure, Pro925 residue corresponds to Pro1008 in Polr2B (RPB2 subunit of POL II). Pro1008 residue is locally involved in a tight turn, where the protein chain changes direction (Figure 2). Substitution p.(Pro925Gln) in POLR3B (RPC2) probably modifies locally the protein conformation, hindering interactions of POLR3B and POLR2L. Indeed, Asp1009 residue in Polr2B (RPB2), corresponding to Asp926 in POLR3B (RPC2), is known to interact with the side chain of Arg48 residue of Polr2l (RPB10), a common subunit of Pol II and Pol III.6, 7 POLR3B and POLR2L interactions are critical for POL III function, since substitution p.(Asp926Glu) in POLR3B has been identified in a 4H-leukodystrophy patient.6 Thus substitution p.(Pro925Gln) probably modifies 3D-position of Asp926 residue, impairing its interaction with POLR2L subunit, leading to the disorder. Variant data have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)—submission ID SUB2188556.

(a) Compound heterozygous mutations c.1568T>A p.(Val523Glu) and c.2774C>A p.(Pro925Gln) were identified in the patient. (b) Mutations c.1568T>A p.(Val523Glu) and c.2774C>A p.(Pro925Gln) induce substitution of highly conserved-across-species residues. (c) Yeast POL II complexed to double-strand DNA (black double helix). RPB2 (Polr2B), highly homologous to POLR3B is colored in red. Pro925 and V523 residues, involved in the substitution, are colored in blue. They correspond to Pro1008 and Leu566 residues in RPB2, respectively. V523 residue is predicted to interact with POLR3D and POLR3E (interaction not shown). Pro925 residue is involved in tight direction change of the protein, which is probably critical for Asp926 interaction with lateral chain of POLR2L (RPB10). The structure and positions of substitutions are illustrated by RasMol (provided courtesy of Bernstein + Sons; http://www.openrasmol.org/) with the crystal structure (PDB accession number 3GTP). The full colour version of this figure is available at European Journal of Human Genetics online.
The c.1568T>A p.(Val523Glu) variant has been identified in 51/62 (82%) of the POLR3B-mutated 4H-leukodystrophy patients.4 Substitution p.(Val523Glu) is predicted to affect POLR3B structure locally, thus impairing POLR3D and POLR3E interaction.8
In order to replicate this finding, we contacted the authors who described earlier four patients with CHES. Unfortunately, patients were lost to follow-up and no samples were stored (Dr C Stoll and Charrow, personal communications).
Discussion
Before the identification of POLR3A, POLR3B and POLR1C as disease-causing genes, five markedly overlapping clinical phenotypes were initially described as distinct entities: 4H, ADDH (ataxia, delayed dentition and hypomyelination), TACH (tremor-ataxia with central hypomyelination), LO (leukodystrophy with oligodontia) and HCAHC (hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum) syndromes. Because of overlapping phenotypes and common molecular basis, these conditions are now thought to belong to the same entity, named POLR3-related leukodystrophies or 4H-leukodystrophy.6, 8, 9, 10, 11, 12 Patients harbor a wide spectrum of severity. Disease onset ranges from neonatal period to late childhood.4, 13 Failure to thrive, hypotonia and developmental delay can be noticed since first year of life. Cerebellar signs become progressively obvious, slowly worsening toward severe intention tremor, dysarthria, dysmetria and gait ataxia. However ataxia progression is variable and thus, cerebellar signs can remain stable over several years. In some patients, variable degree of pyramidal signs and dystonia can occur, worsening further gait disturbance. Patients usually have intellectual disability ranging from mild to severe, gradually worsening in the second decade. Some patients have normal intelligence in early childhood but develop mild intellectual disability during the course of the disease.4 Brain MRI typically shows cerebellar atrophy, thin corpus callosum and hypomyelination associated with relative T2 hypointensity of the ventrolateral thalamus, globus pallidus, optic radiation, internal capsule and dentate nucleus. Patients show also extraneurologic symptoms, consisting in hypodontia with abnormal tooth eruption shape, hypogonadotropic hypogonadism, myopia and short stature.
Recently, POLR3A and POLR3B variants have been identified in patients without diffuse hypomyelination, and even in patients with isolated hypogonadotropic hypogonadism. To date, the latter phenotype represents the milder end of the POLR3-related disorder spectrum.14 CHES is a POLR3-related disorder and represents a severe form of 4H-leukodystrophy. Neurologic impairment starts in the neonatal period with marked hypotonia, global developmental delay and congenital ataxia. Cerebellar signs rapidly worsen hindering independent walking. Intellectual disability is usually mild to severe. Brain MRI, performed only in two patients, show classical MRI phenotype of 4H-leukodystrophy, with diffuse hypomyelination and variable degree of cerebellar atrophy. Patients show classical extraneurologic features, characterized by hypodontia with delayed tooth eruption, myopia and, in our patient, hypogonadotropic hypogonadism. Their skeletal phenotype is striking. All patients have short stature with delayed bone age and endosteal sclerosis. Increased bone density affects particularly pelvis, vertebrae and long bones, causing medullary cavity reduction. Conversely, osteosclerosis is a feature rarely observed in 4H-leukodystrophy, probably because rarely explored. Only three patients were reported with this anomaly, among whom the patient reported by Ozglen et al4 (Table 1). It probably suggests that the skeletal phenotype is a new, previously unrecognized, feature of 4H-leukodystrophy spectrum. Thus, in addition to the classical extraneurologic features, skeletal features and hip dislocation should be carefully looked for, in all patients with POLR3-related disorder.
Molecular finding in CHES patients do not allow establishment of genotype–phenotype correlations, to explain why CHES have a severe neurological and skeletal phenotype. Both CHES patients share the recurrent c.1568T>A p.(Val523Glu) POLR3B variant, also found in about 82% of 4H-leukodystrophy with molecular anomalies of POLR3B.4 They also carry the c.303+1G>A p.(?) and c.2774C>A p.(Pro925Gln) variants, located in intron 5 and exon 26, inducing splicing anomaly and Pro925Gln substitution, respectively. However, Wolf et al4 showed that 4H-leukodystrophy patients with POLR3B variants compared to POLR3A have slightly earlier disease onset and more severe motor involvement with earlier loss of supported walking and higher prevalence of non-ambulatory patients. Our findings strengthen these observations, sorting POLR3B-mutated patients at the severe end of POLR3-related phenotypic spectrum.
References
Stoll C, Talon P, Alembik Y, Levy JM : Congenital cerebellar hypoplasia with bone lesions. Ann Pediatr 1986; 33: 417–421.
Charrow J, Poznanski AK, Unger FM, Robinow M : Autosomal recessive cerebellar hypoplasia and endosteal sclerosis: a newly recognized syndrome. Am J Med Genet 1991; 41: 464–468.
Ozgen HM, Overweg-Plandsoen WC, Blees-Pelk J, Besselaar PP, Hennekam RC : Cerebellar hypoplasia-endosteal sclerosis: a long term follow-up. Am J Med Genet A 2005; 134A: 215–219.
Wolf NI, Vanderver A, van Spaendonk RM et al: Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014; 83: 1898–1905.
Thiffault I, Wolf NI, Forget D et al: Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015; 6: 7623.
Saitsu H, Osaka H, Sasaki M et al: Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011; 89: 644–651.
Wang Z, Roeder RG : Three human RNA polymerase III-specific subunits form a subcomplex with a selective function in specific transcription initiation. Genes Dev 1997; 11: 1315–1326.
Tetreault M, Choquet K, Orcesi S et al: Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet 2011; 89: 652–655.
Hennekam RC : What to call a syndrome. Am J Med Genet A 2007; 143A: 1021–1024.
Wolff A, Koch MJ, Benzinger S et al: Rare dental peculiarities associated with the hypomyelinating leukoencephalopathy 4H syndrome/ADDH. Pediatr Dent 2010; 32: 386–392.
Bernard G, Chouery E, Putorti ML et al: Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 2011; 89: 415–423.
Potic A, Brais B, Choquet K, Schiffmann R, Bernard G : 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol 2012; 69: 920–923.
La Piana R, Cayami FK, Tran LT et al: Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology 2016; 86: 1622–1626.
Richards MR, Plummer L, Chan YM et al: Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genet 2017; 54: 19–25.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Ghoumid, J., Petit, F., Boute-Benejean, O. et al. Cerebellar hypoplasia with endosteal sclerosis is a POLR3-related disorder. Eur J Hum Genet 25, 1011–1014 (2017). https://doi.org/10.1038/ejhg.2017.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.73
