
Abstract
Over 150 different proteins attach to the plasma membrane using glycosylphosphatidylinositol (GPI) anchors. Mutations in 18 genes that encode components of GPI-anchor biogenesis result in a phenotypic spectrum that includes learning disability, epilepsy, microcephaly, congenital malformations and mild dysmorphic features. To determine the incidence of GPI-anchor defects, we analysed the exome data from 4293 parent–child trios recruited to the Deciphering Developmental Disorders (DDD) study. All probands recruited had a neurodevelopmental disorder. We searched for variants in 31 genes linked to GPI-anchor biogenesis and detected rare biallelic variants in PGAP3, PIGN, PIGT (n=2), PIGO and PIGL, providing a likely diagnosis for six families. In five families, the variants were in a compound heterozygous configuration while in a consanguineous Afghani kindred, a homozygous c.709G>C; p.(E237Q) variant in PIGT was identified within 10–12 Mb of autozygosity. Validation and segregation analysis was performed using Sanger sequencing. Across the six families, five siblings were available for testing and in all cases variants co-segregated consistent with them being causative. In four families, abnormal alkaline phosphatase results were observed in the direction expected. FACS analysis of knockout HEK293 cells that had been transfected with wild-type or mutant cDNA constructs demonstrated that the variants in PIGN, PIGT and PIGO all led to reduced activity. Splicing assays, performed using leucocyte RNA, showed that a c.336-2A>G variant in PIGL resulted in exon skipping and p.D113fs*2. Our results strengthen recently reported disease associations, suggest that defective GPI-anchor biogenesis may explain ~0.15% of individuals with developmental disorders and highlight the benefits of data sharing.
Similar content being viewed by others


Introduction
In mammalian cells, there are thought to be over 150 different proteins that are attached to the plasma membrane using a glycosylphosphatidylinositol (GPI) anchor. This diverse family comprises receptors, adhesion molecules and enzymes and is critical for normal neuronal and embryonic development. The GPI anchor is synthesised and remodelled in a complex series of biochemical reactions that take place either in the endoplasmic reticulum (ER) or Golgi apparatus, and at least 30 genes are known that encode components of this pathway.1, 2
The clinical significance of this pathway was first demonstrated in 1993 when somatic mutations in PIGA (which encodes subunit A of phosphatidylinositol N-acetylglucosaminyltransferase) were shown to cause paroxysmal nocturnal haemoglobinuria.3 This rare life-threatening disease results from complement-mediated haemolysis due to a deficiency of surface expression of GPI-anchored complement inhibitors CD55 and CD59. At the time, it was speculated that constitutive mutations in this gene would be embryonically lethal, however, this turned out not to be the case and several overlapping phenotypes have now been associated with germline variants.4, 5, 6, 7, 8
In 2014, using a combination of exome and targeted gene sequencing, we identified three families where individuals with learning disability and hyperphosphatasia harboured biallelic mutations in PGAP3.9 Our work, together with results from many other research groups worldwide, have suggested disease associations for at least 18 genes that relate to GPI-anchor biosynthesis (Supplementary Table S1) and the importance of testing this pathway in clinical diagnostics is now increasingly recognised.2
Although the phenotype associated with GPI-defects is variable, global developmental delay is the most consistent finding (Supplementary Table S1).10 Therefore, seeking to replicate our earlier findings, determine the true incidence of GPI defects in a large unbiased cohort and potentially to identify novel disease–gene associations, we analysed data from the Deciphering Developmental Disorders (DDD) study. This project is a collaboration between the Wellcome Trust Sanger Institute and all 24 Regional Genetics Services in the UK and the Republic of Ireland that aims to facilitate the translation of genomic sequencing technologies into the National Health Service. DDD’s analysis of an initial set of 1133 children with severe undiagnosed developmental disorders revealed a genetic variant that is likely to be causative in 317 cases,11 which provides considerable scope for providing diagnoses or identifying novel disease genes in the remaining cases. The study has now identified at least 16 new genes responsible for developmental disorders.12, 13 Although recruitment to this study ceased in April 2015, with more than 14 000 patients enrolled, the DDD study represents one of the largest exome sequencing initiatives in the world.
Materials and methods
Recruitment and patient details
Patient recruitment was undertaken by all Regional Genetics Services in the UK and the Republic of Ireland. Clinical details for the families of interest are summarised in Table 1 and Supplementary Table S2. The DDD study has been described in more detail elsewhere.11, 12, 13 More information about the aims of the project, subject recruitment and a list of publications are available at www.ddduk.org.
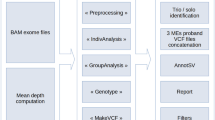
Exome analysis and DDD data filtering
Exome sequencing and bioinformatic methods are described in the Supplementary Methods. Potential candidate variants were identified in individuals using VCF files generated by the DDD study and filtering QC-passed variants as follows:
-
In an initial data set of 1133 trios, the minor allele frequency (MAF) threshold was 1% for all inheritance models. To improve specificity in the expanded data set of 4293 trios, the MAF threshold for monoallelic variants was reduced to 0.1%.
-
Variant effect predictor annotation had to suggest the most severe consequence of the variant is protein altering.
-
Inherited missense alterations predicted benign by PolyPhen-2 were excluded.
-
Genotype and inheritance had to be consistent with a monoallelic mode (de novo or dominantly inherited from affected parent), biallelic mode (homozygous or compound heterozygous) or X-linked mode (hemizygous).
Resulting candidate variants were then filtered for the 31 genes listed in Supplementary Table S1. For trios of interest, a list of all candidate variants was provided. Additional genetic information available included full v4.1 VCFs, annotation for variants that have already been reported back to clinicians via DECIPHER14 and a list of Sanger validated de novo mutations called by DeNovoGear.15 Selected BAM files were downloaded from the European Genome-Phenome Archive (EGA; www.ebi.ac.uk/ega/datasets/EGAD00001001114). Other information included clinical details, which included a list of Human Phenome Ontology terms, information about family relationships and contact details for the referring clinician. Additional information such as VCF files and phenotypic data are available at www.ebi.ac.uk/ega/studies/EGAS00001000775 and the diagnostic variants have been made publicly available through the DECIPHER database:
https://decipher.sanger.ac.uk/patient/257982#genotype
https://decipher.sanger.ac.uk/patient/259633#genotype
https://decipher.sanger.ac.uk/patient/258094#genotype
https://decipher.sanger.ac.uk/patient/270250#genotype
https://decipher.sanger.ac.uk/patient/270306#genotype
https://decipher.sanger.ac.uk/patient/263039#genotype
https://decipher.sanger.ac.uk/patient/277013#genotype
Re-analysis with alternative genome analysis pipeline
It is well known that there is a low genotype concordance between different variant calling software.16 Therefore, data from three families where BAM files were available in EGA were re-analysed with an analysis pipeline that combined multi-sample variant calling with Platypus17 and variant prioritisation using Ingenuity Variant Analysis (www.ingenuity.com/products/variant-analysis), similar to that described previously.18 For three families where BAM files were not available in EGA at the time of the analysis, we uploaded the VCF files that had been generated from the DDD pipeline to Ingenuity Variant Analysis. We filtered variants looking for both de novo and recessive candidate variants using a variety of settings to help confirm that the GPI pathway variants that came up from the primary analysis were the most likely candidates. Read alignments supporting variants of interest were also viewed using the Integrative Genomics Viewer (www.broadinstitute.org/igv).
Sanger validation
The genomic loci surrounding each of the putative pathogenic variants were PCR amplified using the primers listed in Supplementary Table S3. PCRs were purified using standard methods and bidirectional Sanger sequencing was performed using BigDye chemistry (Applied Biosystems, Foster City, CA, USA).
Functional analysis of PIGN, PIGT and PIGO variants
PIGN-knockout HEK293 cells were generated and transfected as described previously,19 with human wild-type or p.(L311W) mutant PIGN cDNA cloned into pME, a strong SRα promoter-driven expression vector, or pTK, a medium TK promoter-driven expression vector. PIGN constructs had an HA epitope tag at the N terminus. After 3 days, restoration of the cell surface expression of CD59 was evaluated by flow cytometry. The strong promoter is useful for detecting complete LoF and severe partial LoF, while the medium promoter is helpful for detecting mild partial LoF because overexpression of mild partial LoF mutant often causes full restoration of CD59.
Levels of expressed wild-type and p.(L311W) mutant HA-tagged PIGN in pME-vector transfected cells were analysed by western blotting using an anti-HA antibody (C29F4, Cell Signaling Tec, Danvers, MA, USA). Levels of protein expression were normalised by the luciferase activity for transfection efficiencies and by expression levels of GAPDH for loading controls.
PIGT and PIGO-knockout HEK293 cells were generated by CRISPR/Cas system and the corresponding PIGT and PIGO variants were assessed by measuring the restoration of CD59 surface expression. Western blotting was used to analyse the protein levels. These experiments were performed as described for PIGN, except PIGT cDNA constructs were FLAG-tagged at the C-terminal and probed with anti-FLAG antibody (M2, Sigma-Aldrich, Saint Louis, MO, USA).
Autozygosity analysis and calculation of inbreeding coefficients
Allelic ratios from a set of high-quality variants were extracted as described in the Supplementary Methods. These data were loaded into Nexus CN (BioDiscovery, El Segundo, CA, USA) to call cnLOH segments across the whole genome. We estimated the coefficient of inbreeding as the total fraction of the autosomal genome, which appeared to be homozygous by descent.
RNA analysis of PIGL splice variant
Fresh blood was collected into PAXgene Blood RNA Tubes and RNA extractions were performed with the PAXGene Blood RNA kit (Qiagen, Manchester, UK). cDNA was reverse transcribed using the QuantiTect kit (Qiagen) and a mixture of oligo-dT and random primers. Forward primers were designed in exons 1 and 2 while reverse primers were designed in exons 5 and 6 (Supplementary Table S3). RT-PCR products were diluted and run on a High Sensitivity DNA Chip on the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). PCR products were also purified using exoI (NEB, Ipswich, MA, USA) and shrimp alkaline phosphatase (USB, Cleveland, OH, USA) and Sanger sequencing was performed as described above.
Results
Summary of candidates and exclusion criteria
The DDD filtering pipeline identified 43 patient–parent trios (42 independent families and two siblings) in which rare, potentially functional candidate variants were identified in at least one of the GPI-anchor biogenesis genes. As has been noted previously,11 parental-affected status significantly influenced the number of candidate variants identified. Across the entire exome, there were on average 65.8 candidate variants in trios where both parents were affected (mostly variants inherited from one or other parent), 34.1 candidates where just a single parent was affected and just 6.7 candidates (range 2–16) where neither parent was affected.
As of July 2015, four of the 43 index cases had variants in other (ie, non-GPI pathway) genes reported that were already considered to be clinically relevant. For instance, a girl with developmental delay and ASD (DECIPHER ID 258536) harboured a de novo p.(Q1093*) mutation in SYNGAP1 (NM_006772.2).20
GPI-anchor biogenesis genes reported to date (Supplementary Table S1) are all associated with recessively inherited conditions. We therefore focussed on variants that fitted a biallelic inheritance (ie, compound heterozygous or homozygous) or X-linked recessive models, excluding families where parents were affected and candidate variants fitted a dominant inheritance model.
Focussing on a recessive model also led us to ignore putative de novo missense variants in PIGM (c.1199A>G; p.(N400S), NM_145167.2) and MPPE1 (c.682C>T; p.(R228C), NM_023075.5). The inheritance pattern associated with PIGM mutations has been reported to be autosomal recessive.21 We also note that both these genes have low pLI scores in ExAC v0.3 and so are unlikely to be sensitive to haploinsufficiency.22 After further review of candidates, we also excluded a small set of variants, which were detected at MAF 0.1–1.0% but were each present in a homozygous state in ExAC V0.3 multiple times. This led us to exclude patients with biallellic variants in PIGW (c.705C>G;705C>G in individual 275308, c.705C>G;908G>A in 259553, NM_178517.3), PIGS (c.553C>T;553C>T in 267380, NM_033198.3) and GPLD1 (c.308A>G;2442delA in 276507, NM_001503.3).
Overview of likely causative variants
As a result of the above filtering, potentially clinically significant variants were identified in 7/4293 parent–child trios. These consisted of 11 rare variants spread across five different GPI-anchor biogenesis genes (Figure 1). In five of the families, the variants were in a compound heterozygous configuration. The sixth family was a consanguineous Afghani kindred with two affected brothers and here the likely causative mutation was homozygous.

Pedigrees and genetic data for six families harbouring rare biallelic variants in genes encoding components of the GPI-anchor biogenesis pathway. The Sanger sequencing traces shown are for the proband in each family and are shown in the coding direction, alongside the corresponding wild-type amino acid sequence. In the case of PIGT family 2 we show a trace from the father, where the variant is in the heterozygous state. For PIGT family 1 and the PIGL family, DNA was not available for the unaffected older siblings. Codon numbering is with respect to the following GenBank transcripts; PGAP3: NM_033419.4; PIGN: NM_176787.4; PIGT: NM_015937.5; PIGO: NM_032634.3; PIGL: NM_004278.3.
Including the Afghani quartet, DNA from affected or unaffected siblings was available for testing in four out of six of the families and in all cases, the segregation pattern was consistent with the variants being causative (Figure 1; P=0.026). For four out of five genes where alkaline phosphatase testing is known to be informative, abnormal results were obtained and the directionality was as expected, that is, elevated with mutations in three out of five genes, normal with one out of five genes and lowered or close to lower limit with mutations in one out of five genes (Table 1). None of the variants were reported to occur in a homozygous state in ExAC, with total allele counts ranging from 0 to 16 (Table 1).
PGAP3 family
Individual 257982 harboured rare compound heterozygous variants in PGAP3: a c.914A>G (predicting a p.(D305G) alteration to the amino acid sequence) inherited from the patient’s father and a c.320C>T change (predicting p.(S107L)) from the mother. We note that p.(D305G) was described previously (family B in Howard et al.9) where it was shown to result in abnormal protein localisation to the ER. p.(S107L) was identified in a more recent study where it was shown to reduce PGAP3 activity.23 In one case (family D in Knauss et al.23), the same two variants were identified as in 257982. However, in that patient, p.(S107L) was paternal and p.(D305G) maternal.
Sanger sequencing confirmed that both variants were present in the affected brother of 257982 (Figure 1). In both affected siblings, alkaline phosphatase activity was increased (Table 1), consistent with the results reported previously.9
PIGN family
Individual 259633 harboured compound heterozygous variants in PIGN: a c.932T>G (predicting p.(L311W)) from the father and a c.694A>T (predicting p.(K232*)) from the mother. Sanger sequencing of two unaffected siblings indicated that neither had inherited both variants (Figure 1). Both variants have been described recently; a homozygous p.(K232*) mutation was seen in a fetus diagnosed with Fryns syndrome,24 a condition characterised by multiple congenital anomalies, while p.(L311W) was observed in an individual where the phenotype was limited to hypotonia, developmental delay and seizures.25
Alkaline phosphatase testing for this case is uninformative as normal results are expected for patients with PIGN mutations19, 26 and therefore functional assessment was performed using PIGN-knockout HEK293 cells. With an expression plasmid using a strong pME promoter, a wild-type PIGN restored CD59 expression on 52% of PIGN-knockout cells after transient transfection, whereas p.(L311W) PIGN restored CD59 on only 39% of the cells (Figure 2a, left panel). With a medium promoter plasmid pTK, the wild-type PIGN restored CD59 on a small fraction of the cells, whereas the p.(L311W) PIGN had almost no effect (right panel). Western blot analysis indicated that the missense alteration did not significantly affect protein expression (Figure 2b). These results indicate that the p.(L311W) mutation reduces enzymatic activity rather than affecting protein levels.

Follow-up studies on variants in PIGN and PIGT. (a) PIGN-knockout HEK293 cells were generated and transfected with human wild-type or p.(L311W) mutant PIGN cDNA cloned into pME or pTK expression vectors. Restoration of the cell surface expression of CD59 was evaluated by flow cytometry. The mutant construct using the pME promoter did not rescue CD59 surface expression as efficiently as the wild-type construct, indicating that the variant results in reduced PIGN activity. (b) Levels of expressed wild-type and p.(L311W) mutant HA-tagged PIGN in pME-vector transfected cells were analysed by western blotting using an anti-HA antibody. After normalisation with luciferase activity and GAPDH, expression of the mutant protein appeared to be reduced by only ~10% compared with the wild-type protein. (c) PIGT-knockout HEK293 cells were transfected with wild-type or mutant PIGT cDNA cloned into pME or pTK expression vectors. Restoration of the cell surface expression of CD59 was evaluated by flow cytometry. The mutant constructs using the pTK promoter did not rescue CD59 surface expression as efficiently as the wild-type construct, indicating that the variants result in reduced PIGT activity. (d) Levels of expressed wild-type and mutant FLAG-tagged PIGT in pME-vector transfected cells were analysed by western blotting. After normalisation, expression of the mutant protein appeared to be reduced only for the p.(L578fs*35) variant. (e) Allelic ratio plots along chromosome 20 (for high confidence SNVs only) showed that the PIGT variant shared in 270250 and 270306 lies within a large region of autozygosity.
PIGT family 1
Individual 258094 harboured compound heterozygous variants in PIGT: c.1582G>A (predicting p.(V528M)) from the mother and c.1730dupC (predicting p.(L578fs*35)) from the father. Sanger sequencing was used to validate both variants, although DNA from the unaffected sister was unavailable for testing. Initial publications on this gene reported decreased alkaline phosphatase activity27, 28 but a subsequent study found normal levels.29 In this case, alkaline phosphatase activity was in the normal range (Table 1). Rescue experiments performed on PIGT-knockout HEK293 cells indicated that both mutations result in a mild reduction in the amount of CD59 anchored to the cell membrane, although this effect was only seen when using the pTK promoter (Figure 2c). Western blot analysis suggested that p.(L578fs*35) may lead to a small decrease in protein stability (Figure 2d). The functional effect of these two mutations was further confirmed by the reduced CD16 expression seen on patient granulocytes (Supplementary Figure S1).
Recent studies have shown complex multisystem conditions can be a result from blending of two distinct genetic disorders.30, 31, 32, 33 In that respect, we note that 258094 also harboured compound heterozygous variants in PKHD1 (predicting p.(P2319Q); p.(D3923fs*8), NM_138694.3). This gene is associated with Autosomal Recessive Polycystic Kidney Disease (AR-PKD), a severe condition in which a significant fraction of babies die within the first 4 weeks of life due to breathing difficulties. Although 258094 had kidney stones, nephrolithiasis is not typically a feature of AR-PKD.
PIGT family 2
Individual 270250 harboured a homozygous c.709G>C variant (predicting p.(E237Q)) in PIGT. An affected brother (270306) was confirmed by both Sanger sequencing and exome analysis to be homozygous for the same variant (Figure 1). Alkaline phosphatase activity for 270250 was below the normal range, whereas for the younger brother it was at the lower end of the normal range (Table 1). FACS analysis of PIGT-knockout HEK293 cells showed that p.(E237Q) results in a small reduction in the amount of CD59 anchored to the cell membrane (Figure 2c).
Using allelic ratio information obtained from the exome data, we estimated the coefficients of inbreeding for 270250 and 270306 to be 1/15 and 1/19 respectively, consistent with the 1/16 theoretical expectation for offspring of first-cousin marriages. The PIGT gene was shown to lie within a 10–12 Mb region of autozygosity (Figure 2e). The only larger region of autozygosity shared by both siblings was a 35.5 Mb segment on the short arm of chromosome 2 (data not shown).
PIGO family
Individual 263039 harboured compound heterozygous variants in PIGO: c.1306C>T (predicting p.(R436W)) from the mother and c.713G>A (predicting p.(G238D)) from the father. The unaffected elder brother did not have either variant. Alkaline phosphatase activity was intermittently raised, as is expected.34 FACS analysis of PIGO-knockout HEK293 cells showed that p.(G238D) resulted in no detectable activity, consistent with its position within the Type 1 phosphodiesterase/nucleotide pyrophosphatase/phosphate transferase domain and the conservation of Gly238 in known paralogues (PIGN and PIGG). In contrast, p.(R436W) only resulted in a moderate decrease in the amount of CD59 anchored to the cell membrane (Figure 3a). The difference in functional effects could not be explained by protein stability as both missense variants resulted in only a mild decrease in protein expression (Figure 3b).

Follow-up studies on variants in PIGO and PIGL. (a) PIGO-knockout HEK293 cells were transfected with wild-type, p.(R436W) or p.(G238D) PIGO cDNA. Restoration of the cell surface expression of CD59 was evaluated by flow cytometry. The p.(G238D) variant resulted in no detectable activity when using the pME promoter. For the p.(R436W) variant, reduced CD59 surface expression was only observed when using the pTK promoter. (b) Levels of expressed wild-type and mutant HA-tagged PIGO in pME-vector transfected cells were analysed by western blotting. After normalisation, expression of the mutant protein appeared to be mildly reduced for both missense variants. (c) 2100 Bioanalyser image showing PIGL RT-PCR amplicons using primers positioned in exons 2 and 5. A lower band was observed for 277013 and her father, consistent with skipping of exon 3. The expected sizes were calculated to be 280 bp and 189 bp if exon 3 is missing, which is consistent with the observed sizes given the margin for error reported by the manufacturer. Skipping of a 91 bp exon would lead to a frameshift and premature termination codon, as shown in Supplementary Figure S3.
In addition, an X-linked variant of uncertain significance (c.2683T>A, predicting p.(F895I)) was identified in BCORL1 (NM_021946.4), a transcriptional co-repressor gene. Although this variant is very rare and not present in ExAC, the evidence supporting BCORL1 to be a causative gene for learning disability was limited;35 many of the proposed genes for X-linked learning disability have recently been challenged in light of data from large exome sequencing data sets.36
PIGL family
Individual 277013 harboured compound heterozygous variants in PIGL: c.48G>A (predicting p.(W16*)) from the mother and a c.336-2A>G mutation in the exon 3 consensus splice-acceptor site, from the father. DNA from the unaffected brother was unavailable. Alkaline phosphatase results were not reported in the original clinical description of CHIME syndrome37 but in a subsequent case with PIGL mutations were described to be elevated.38 For 277013, alkaline phosphatase levels were persistently raised (Table 1).
RNA analysis of the splice mutation was complicated by the fact that in all the samples, we observed skipping of exon 5, consistent with the Ensembl annotation ENST00000395844. Although this naturally occurring isoform is predicted to result in a LoF allele (p.A166fs*80), we note that this shorter transcript was observed at relatively low levels when compared with the canonical mRNA (Supplementary Figure S2). In view of this, we did not attempt to analyse the exon 3 splice-acceptor mutation using sequence from the ‘6 R’ RT-PCR primer. The analysis of RT-PCR products using the ‘5 R’ primer demonstrated that the c.336-2A>G mutation resulted in a lower band in both 277013 and in her father (Figure 3c). Sanger sequencing confirmed that this was due to complete skipping of exon 3, predicting a frameshift that results in an aspartic acid to tryptophan alteration followed immediately by a premature stop (p.D113fs*2; Supplementary Figure S3) and therefore likely represents a LoF allele.
Although the stop and splice variants are both seen in ExAC (1/121 332 and 6/121 410, respectively), neither occur in a homozygous state. There were also no other homozygous LoF variants in PIGL within ExAC or another project that searched for rare gene knockouts in a cohort enriched for homozygous alleles.39
Overall clinical comparison
Epilepsy and microcephaly was observed in 5/6 and 3/6 of the families, respectively (Table 1). The photographs of patients (Figures 4a–d and data not shown) highlight a number of common facial similarities, most notably the thin tented upper lips and a broad nasal tip apparent in three out of six of the families. Brachydactyly or brachytelephalangy is present in three out of six of the families. This has been previously reported with GPI mutations. Moderate-to-severe intellectual disability is universal. Some patients were noted to have structural brain anomalies such as cerebral atrophy, cerebellar atrophy and Dandy–Walker variant. Other structural abnormalities seen were cleft palate, aganglionic megacolon and renal cysts. Although not individually common, these anomalies have also been previously described.

Clinical images, shown with parental consent. (a) Photographs of individual 257982 aged 2 years and 8 months and her younger affected brother both showing thin upper lip and short nose with a broad nasal tip. Arrow indicates cleft palate, shown for younger sibling but also present in proband. (b) Photograph of 259633 showing thin tented upper lip and a short nose with a broad nasal tip. (c) Photographs of 258094 showing thin upper lip, nose with broad nasal tip and low-set ears; hands show tapering fingers. (d) Photograph of 263039 showing thin Cupid’s-bow shaped upper lip, brachydactyly with absent fifth fingernail and dystrophic fourth and fifth toenails.
Discussion
In this study, we interrogated exome data from 4293 patient–parent trios, looking for rare biallelic variants in 31 genes related to GPI-anchor biogenesis. Seven individuals (from six independent families) were identified, each referred from different Regional Genetics Services across the UK. As the 4293 patients came from 4125 independent families,40 we therefore estimate incidence of GPI biogenesis defects in this patient group to be ~0.15% (6/4125). Other studies on GPI-anchor biogenesis have typically either used much tighter patient selection criteria41 or else large consanguineous families where genetic mapping is possible.9 This is therefore the first study to estimate the prevalence of such defects in a large unbiased cohort with developmental delay.
Together with other recent studies,23, 42 our study serves to confirm the genotype–phenotype correlation for PGAP3 that we first described in 2014.9 Besides the elevated alkaline phosphatase, the most noticeable features that overlap the five original cases are the broad nasal tip and thin upper lips, which were seen in both 257982 and her younger brother (Figure 4a). Future studies should test whether the distinct craniofacial gestalt make this a clinically recognisable condition. Midline hand movements similar to those described in family A in Howard et al9 were reported in the younger brother. Here, the onset of absence and startle seizures was at age 2 years, whereas in published cases, onset was 1.5–23 years and included tonic–clonic and myoclonic forms of epilepsy.9, 23 Microcephaly was observed in 3 out of 13 published cases9 and in the family described here, a small head size was reported only in the younger brother. Hypotonia was also present in both siblings, consistent with the literature. The p.(D305G) and p.(S107L) mutations have now both been described and so have already been functionally validated.9, 23 p.S107L lies close to two other reported mutations (p.(G92D) and p.(P105R)) and so this region of the gene may represent a hotspot for disease-causing mutations.
As well as confirmation of recently reported genotype–phenotype correlations, our study also helps to delineate the phenotypic range associated with certain GPI-anchor biogenesis genes. For instance, Hirschsprung’s disease (HD), which is a relatively common feature in cases with ‘hyperphosphatasia with mental retardation syndrome’ (HPMRS1) due to PIGV mutations (OMIM #239300),43 has only been reported in one individual with PIGO mutations (HPMRS2; OMIM #614749).44 The HD diagnosis for 263039 therefore provides additional evidence that intestinal disorders can be observed across different genetic HPMRS subtypes. Although seizures were not reported (at 2 years of age), in other respects such as the cupid’s-bow-shaped upper lip, intermittently elevated alkaline phosphatase, hypoplasia of distal phalanges, postnatal microcephaly and hearing loss, the phenotype for 263039 appears to be similar to published cases.34, 44, 45
Mutations in PIGV are thought to represent the major cause of ‘hyperphosphatasia with mental retardation syndrome’43 and so we were surprised that this gene did not come up in our analysis. We therefore investigated the possibility that we were being overly stringent with our MAF filter. The most common PIGV mutation (c.1022C>A; p.(A341E)) is categorised as probably damaging by PolyPhen-2 and present in ~80% of affected families.43 However, in ExAC, this variant is seen at a maximum MAF of 17/66 740 alleles (0.025%; all heterozygous) within the non-Finnish European population, well below not only the initial 1% cut-off for biallelic variants, but also the 0.1% filter that we applied following manual review of variants.
Although this study has helped replicate relatively new disease genes, all five for which the primary disease association was published since 2011 (Table 1), we were unable to identify likely causative variants in any of the 13 genes in the GPI-anchor biogenesis pathway for which disease associations have not yet been reported. It may be that these genetic conditions are so rare that a larger cohort is needed to identify such families. Alternatively, individuals with variants in other GPI genes might not present with developmental delay. For instance, a recent study suggests that mutations in PIGC are embryonically lethal.46
One limitation of this study is that missense alterations predicted benign by PolyPhen-2 would be missed. We also excluded variants, which appeared homozygous multiple times within the ExAC cohort. Although we felt these filters were necessary to improve specificity while analysing such a large cohort, it means that our ~0.15% estimate of incidence may represent an underestimate. We also acknowledge that our use of WES (rather than WGS) would miss deep intronic variants or structural variants such as inversions. In particular, we cannot exclude that the de novo variants in PIGM and MPPE1 occurred in trans with one such variant. Our understanding of GPI-anchor biogenesis in humans may be incomplete. Additional genes involved with this pathway may await discovery and so our candidate gene list should be considered a non-exhaustive list. This could again contribute to an underestimation of the true incidence. Another limitation is that in most cases we were unable to perform FACS analysis to assess levels of GPI-anchored proteins on patient granulocytes, instead relying on phenotypic overlap, segregation testing, alkaline phosphatase activity and functional results from HEK293 cells to accumulate evidence supporting pathogenicity. For all five genes identified, multiple families are already described in the literature. As the phenotypes of the patients described here showed significant overlaps with published cases, we felt that once the variants had been validated, requesting further venepunctures was not warranted. The only exception to this was the girl from PIGT family 1, where alkaline phosphatase results were normal and phenotypic overlap was nonspecific. For this case, FACS analysis of patient granulocytes indicated a mild decrease in surface CD16 levels. For the girl with PIGN variants, the clinical overlap with published cases also showed limited specificity. Biallelic variants in PIGN cause ‘multiple congenital anomalies-hypotonia-seizures syndrome type 1’ (MCAHS1; OMIM 614080).19, 26 However, a recent review of published cases highlights significant phenotypic heterogeneity.24 Although seizures, developmental delay and hypotonia are always present, other features can include dysmorphisms (low-set ears, micrognathia and distal digital hypoplasia), cerebellar atrophy, nystagmus and diaphragmatic hernia. Therefore, although the phenotype observed for individual 259633 (epilepsy, developmental delay, hypotonia and mild brain atrophy) does overlap, we considered the presentation to be nonspecific. In addition, for PIGN mutations, alkaline phosphatase testing is not informative as PIGN deficient individuals do not have hyperphosphatasia. This may be because GPI lacking an EtNP-side branch on Man1 is efficiently added to ALP when GPI transamidase cleaves the GPI-attachment signal sequence.47 Using PIGN-knockout HEK293 cells, we confirmed that p.(L311W) results in reduced PIGN activity. Jezela-Stanek et al.25 recently described a similar case with a relatively mild phenotype (seizures, developmental delay and hypotonia) and reduced expression of GPI-APs in patient granulocytes. It is interesting to note that the p.(L311W) variant is also shared in common between these two milder cases. Although p.(L311W) appears to retain some activity, p.(K232*) in contrast is presumably a LoF allele and this might explain why homozygosity of the p.(K232*) variant resulted in the severe prenatal presentation reported recently by McInerney-Leo et al.24
To facilitate the consistent interpretation of genetic variants between different clinical genetics laboratories, the American College of Medical Genetics and Genomics (ACMG) has developed detailed guidelines about how variants should be interpreted in a systematic way.48 Using this scoring system, we classified the 11 variants described in Figure 1 and note that seven of these variants are scored as pathogenic, whereas for four of the variants there is only enough evidence to reach a ‘likely pathogenic’ classification (Supplementary Table S4). A recent study showed that even following these recommendations, variant scoring can be inconsistent. Although consensus meetings can improve concordance between laboratories, agreement is not always reached for many variants and further clarifications may be beneficial.49 The scoring scheme allows a degree of flexibility and certain criteria can be increased in evidence strength based on expert judgement. For example, both PGAP3 variants described here have now been described in trans with pathogenic variants in three unrelated patients and so the PM3 criteria should be upgraded from moderate to strong. In two cases, we upgraded an inferred classification of ‘likely pathogenic’ to ‘pathogenic’. For instance, although the p.(L311W) variant in PIGN has been described before,25 this was only in a single affected individual and so we could not invoke PS4, which requires multiple prior observations. But together with the modest co-segregation seen in our family (again, not reaching the level to invoke PP1) and the robust functional experiments performed here using mutant HEK293 cells (Figure 2) and by Jezela-Stanek using patient cells, this was enough to persuade us that this variant is pathogenic.
In conclusion, our study suggests that defective GPI-anchor biogenesis may explain ~0.15% of cases with developmental delay and increases the yield of clinically relevant findings within the DDD patient group that are available for families to help with recurrence risk counselling and potentially the provision of further genetic testing. The results also help confirm and extend the phenotypic range of recently reported disease genes and exemplify the benefits of large scale data sharing, providing a model for other large genomic projects such as the UK’s 100 K genomes project.
References
Kinoshita T : Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proc Jpn Acad Ser B Phys Biol Sci 2014; 90: 130–143.
Ng BG, Freeze HH : Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). J Inherit Metab Dis 2015; 38: 171–178.
Takeda J, Miyata T, Kawagoe K et al: Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 1993; 73: 703–711.
Belet S, Fieremans N, Yuan X et al: Early frameshift mutation in PIGA identified in a large XLID family without neonatal lethality. Hum Mutat 2014; 35: 350–355.
Johnston JJ, Gropman AL, Sapp JC et al: The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012; 90: 295–300.
Kato M, Saitsu H, Murakami Y et al: PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology 2014; 82: 1587–1596.
Swoboda KJ, Margraf RL, Carey JC et al: A novel germline PIGA mutation in Ferro-Cerebro-Cutaneous syndrome: a neurodegenerative X-linked epileptic encephalopathy with systemic iron-overload. Am J Med Genet A 2014; 164A: 17–28.
Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S et al: The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis 2015; 10: 23.
Howard MF, Murakami Y, Pagnamenta AT et al: Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am J Hum Genet 2014; 94: 278–287.
Makrythanasis P, Kato M, Zaki MS et al: Pathogenic variants in PIGG cause intellectual disability with seizures and hypotonia. Am J Hum Genet 2016; 98: 615–626.
Wright CF, Fitzgerald TW, Jones WD et al: Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015; 385: 1305–1314.
Akawi N, McRae J, Ansari M et al: Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4125 families. Nat Genet 2015; 47: 1363–1369.
DDD: Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015; 519: 223–228.
Firth HV, Richards SM, Bevan AP et al: DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009; 84: 524–533.
Ramu A, Noordam MJ, Schwartz RS et al: DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods 2013; 10: 985–987.
Gezsi A, Bolgar B, Marx P, Sarkozy P, Szalai C, Antal P : VariantMetaCaller: automated fusion of variant calling pipelines for quantitative, precision-based filtering. BMC Genomics 2015; 16: 875.
Rimmer A, Phan H, Mathieson I et al: Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 2014; 46: 912–918.
Pagnamenta AT, Howard MF, Wisniewski E et al: Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum Mol Genet 2015; 24: 3732–3741.
Ohba C, Okamoto N, Murakami Y et al: PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics 2014; 15: 85–92.
Parker MJ, Fryer AE, Shears DJ et al: De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Med Genet A 2015; 167A: 2231–2237.
Almeida AM, Murakami Y, Layton DM et al: Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 2006; 12: 846–851.
Lek M, Karczewski KJ, Minikel EV et al: Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291.
Knaus A, Awaya T, Helbig I et al: Rare non-coding mutations extend the mutational spectrum in the PGAP3 subtype of hyperphosphatasia with mental retardation syndrome. Hum Mutat 2016; 37: 737–744.
McInerney-Leo AM, Harris JE, Gattas M et al: Fryns syndrome associated with recessive mutations in PIGN in two separate families. Hum Mutat 2016; 37: 695–702.
Jezela-Stanek A, Ciara E, Piekutowska-Abramczuk D et al: Congenital disorder of glycosylphosphatidylinositol (GPI)-anchor biosynthesis—The phenotype of two patients with novel mutations in the PIGN and PGAP2 genes. Eur J Paediatr Neurol 2016; 20: 462–473.
Maydan G, Noyman I, Har-Zahav A et al: Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 2011; 48: 383–389.
Kvarnung M, Nilsson D, Lindstrand A et al: A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 2013; 50: 521–528.
Nakashima M, Kashii H, Murakami Y et al: Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics 2014; 15: 193–200.
Lam C, Golas GA, Davids M et al: Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol Genet Metab 2015; 115: 128–140.
Li Y, Salfelder A, Schwab KO et al: Against all odds: blended phenotypes of three single-gene defects. Eur J Hum Genet 2016; 24: 1274–1279.
Tarailo-Graovac M, Shyr C, Ross CJ et al: Exome sequencing and the management of neurometabolic disorders. N Engl J Med 2016; 374: 2246–2255.
Yang Y, Muzny DM, Reid JG et al: Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013; 369: 1502–1511.
Yang Y, Muzny DM, Xia F et al: Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014; 312: 1870–1879.
Krawitz PM, Murakami Y, Hecht J et al: Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 2012; 91: 146–151.
Schuurs-Hoeijmakers JH, Vulto-van Silfhout AT, Vissers LE et al: Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet 2013; 50: 802–811.
Piton A, Redin C, Mandel JL : XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet 2013; 93: 368–383.
Ng BG, Hackmann K, Jones MA et al: Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 2012; 90: 685–688.
Fujiwara I, Murakami Y, Niihori T et al: Mutations in PIGL in a patient with Mabry syndrome. Am J Med Genet A 2015; 167 A: 777–785.
Narasimhan VM, Hunt KA, Mason D et al: Health and population effects of rare gene knockouts in adult humans with related parents. Science 2016; 352: 474–477.
McRae JF, Clayton S, Fitzgerald TW et al: Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017; 542: 433–438.
Krawitz PM, Murakami Y, Riess A et al: PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet 2013; 92: 584–589.
Yavarna T, Al-Dewik N, Al-Mureikhi M et al: High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet 2015; 134: 967–980.
Horn D, Wieczorek D, Metcalfe K et al: Delineation of PIGV mutation spectrum and associated phenotypes in hyperphosphatasia with mental retardation syndrome. Eur J Hum Genet 2014; 22: 762–767.
Kuki I, Takahashi Y, Okazaki S et al: Vitamin B6-responsive epilepsy due to inherited GPI deficiency. Neurology 2013; 81: 1467–1469.
Nakamura K, Osaka H, Murakami Y et al: PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia 2014; 55: e13–e17.
Shamseldin HE, Tulbah M, Kurdi W et al: Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol 2015; 16: 116.
Hong Y, Maeda Y, Watanabe R et al: Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. J Biol Chem 1999; 274: 35099–35106.
Richards S, Aziz N, Bale S et al: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424.
Amendola LM, Jarvik GP, Leo MC et al: Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet 2016; 98: 1067–1076.
Acknowledgements
We thank the families who kindly agreed to participate in this study, the Wessex regional genetic laboratory for validation of the variants in PIGT family 1, CentoGene for sharing validation and co-segregation data for the PIGO variants and the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) and the groups that provided exome variant data for comparison. We also thank Kana Miyanagi and Saori Umeshita for their technical assistance. This work was supported by the Wellcome Trust (090532/Z/09/Z) and the National Institute for Health Research (NIHR) Biomedical Research Centre Oxford with funding from the Department of Health’s NIHR Biomedical Research Centre’s funding scheme. Additional funding for this study was by grants from the Japanese Agency for Medical Research and Development, AMED and the Ministry of Health Labour and Welfare. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [HICF-1009-003], a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute [WT098051]. The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. The views expressed in this publication are those of the authors and not necessarily those of the Wellcome Trust or the Department of Health.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pagnamenta, A., Murakami, Y., Taylor, J. et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet 25, 669–679 (2017). https://doi.org/10.1038/ejhg.2017.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.32
This article is cited by
-
A novel homozygous PIGO mutation associated with severe infantile epileptic encephalopathy, profound developmental delay and psychomotor retardation: structural and 3D modelling investigations and genotype–phenotype correlation
Metabolic Brain Disease (2023)
-
Molecular insights into biogenesis of glycosylphosphatidylinositol anchor proteins
Nature Communications (2022)
-
Assessing reproducibility of inherited variants detected with short-read whole genome sequencing
Genome Biology (2022)
-
Loss of PIGK function causes severe infantile encephalopathy and extensive neuronal apoptosis
Human Genetics (2021)
-
Analyzing clinical and genetic characteristics of a cohort with multiple congenital anomalies-hypotonia-seizures syndrome (MCAHS)
Orphanet Journal of Rare Diseases (2020)
