
Abstract
Variants in the ZIC3 gene are rare, but have demonstrated their profound clinical significance in X-linked heterotaxy, affecting in particular male patients with abnormal arrangement of thoracic and visceral organs. Several reports have shown relevance of ZIC3 gene variants in both familial and sporadic cases and with a predominance of mutations detected in zinc-finger domains. No studies so far have assessed the functional consequences of ZIC3 variants in an in vivo model organism. A study population of 348 patients collected over more than 10 years with a large variety of congenital heart disease including heterotaxy was screened for variants in the ZIC3 gene. Functional effects of three variants were assessed both in vitro and in vivo in the zebrafish. We identified six novel pathogenic variants (1,7%), all in either male patients with heterotaxy (n=5) or a female patient with multiple male deaths due to heterotaxy in the family (n=1). All variants were located within the zinc-finger domains or leading to a truncation before these domains. Truncating variants showed abnormal trafficking of mutated ZIC3 proteins, whereas the missense variant showed normal trafficking. Overexpression of wild-type and mutated ZIC protein in zebrafish showed full non-functionality of the two frame-shift variants and partial activity of the missense variant compared with wild-type, further underscoring the pathogenic character of these variants. Concluding, we greatly expanded the number of causative variants in ZIC3 and delineated the functional effects of three variants using in vitro and in vivo model systems.
Similar content being viewed by others


Introduction
Congenital heart disease (CHD) is one of the most frequent malformations at birth, with an estimated incidence of one in 100–250 live births worldwide.1 Fortunately for patients, thanks to great improvements in both medical and surgical treatment, survival rates of CHD patients have now increased to >90%.2 The clinical spectrum of CHD is very broad, encompassing a large group of both structural as well as functional anomalies arising in early cardiac development.3 This diversity is mirrored by the complex etiology of CHD with both environmental as well as many genetic factors being implicated in different or even identical cardiac defects.4 A very small subset within the spectrum of CHD’s known as heterotaxy syndrome occurs in approximately one in 5000–7000 live births.5 This syndrome is defined by a variety of both cardiac and non-cardiac congenital malformations induced by errors of the left-right axis determination in early embryonic development. As the normal heart is one of the most asymmetric organs in the body, failure to correctly establish this asymmetry can result in mixing of oxygenated and deoxygenated blood, creating cyanotic heart disease just after birth.6, 7 Although the major morbidity in heterotaxy is mostly the result of complex cardiac malformations, the syndrome is also characterized by abnormal arrangements of other internal organs, including lungs, spleen, liver, stomach and intestine.5, 8 Heterotaxy occurs in the majority of patients as a sporadic condition but clustering in families has been documented, indicating several modes of inheritance including autosomal dominant,9, 10 autosomal recessive11 and X-linked.12 In the past two decades several genes were implicated in heterotaxy in a minority of patients such as ZIC3,13 NODAL,14 CFC1,15 ACVR2B,16 GDF1,17 SHROOM318 and CFAP53.19 Variants in the ZIC3 gene, located on Xq26.2 (HTX1, MIM 306955) cause X-linked heterotaxy.13 The ZIC3 gene is a C2H2 zinc-finger transcription factor belonging to the GLI superfamily involved in neurogenesis. Humans, frog, chicken and mice have five ZIC homologs and the zebrafish has seven, all with very conserved DNA-binding zinc-finger motifs.20 The evolutionary conservation of these genes and their critical timing of expression is essential for proper embryonic development, since variants in other human zic genes also cause monogenic diseases, for instance ZIC2 causing holoprosencephaly.21 The ZIC3 gene, however, is the only ZIC gene to be implicated in laterality disorders. Several animal studies have demonstrated the crucial role of ZIC3 expression during gastrulation and axial patterning and the laterality abnormalities created when knocked out.22, 23 The first publication on pathogenic variants in ZIC3 causing X-linked heterotaxy dates more than 15 years ago13 and until now the number of reports on ZIC3 variants are limited. Even though this may indicate that the incidence is very low, studies have suggested that patients with only specific cardiac lesions and no laterality defects may also carry pathogenic ZIC3 variants.24
In this study, we show the results of ZIC3 gene screening in a group of 348 patients collected over a time period of 10 years. The group of patients included heterotaxy patients as well as patients with a large variety of CHD. We identified six novel pathogenic variants, increasing the total number reported of distinct pathogenic variants from 30 to 36. We also performed both in vitro and in vivo functional testing of three of the novel variants in cell culture and the zebrafish respectively, further extending our knowledge of functional characteristics.
Materials and Methods
Patient cohort
Between 2003 and 2014 patients were referred to the Department of Clinical Genetics for ZIC3 genetic testing. For this study the patients were categorized in the following situs subgroups: classic heterotaxy (combination of complex cardiac malformations and disrupted laterality of at least one organ), CHD heterotaxy (normal situs of other organs than the heart), abdominal heterotaxy (normal heart and at least one abdominal organ with disrupted laterality), situs inversus totalis and situs solitus with solely heart defects (subdivided in complex CHD (more than one cardiac defect), septation defects (atrial septum defect (ASD), ventricular septum defect (VSD) or artioventricular septum defect (AVSD)), left-sided obstruction defects (hypoplastic left heart syndrome (HLHS)), cyanotic heart disease (transposition of the great arteries (TGA), tetralogy of Fallot (TOF)) or other (bicuspid aortic valve (BAV), coarctatio aortae (CoA), pulmonary stenosis (PS), Ebstein anomaly)). Heterotaxy was considered 'familial' if at least one other family member also showed any form of laterality disorder. Informed consent was obtained from all patients (or parents) and family members, in accordance with guidelines of the local ethical committee. If applicable, consent for the publication of photographs was also obtained.
Variant analysis and classification
Blood samples, cultured fibroblasts or amniotic fluid cells were used for DNA extraction following standard procedures. In addition, blood samples from family members were obtained when possible. Genetic analysis of the complete coding region, including intron–exon boundaries, of the ZIC3 gene (Xq26.2, HTX1, OMIM #306955) was performed by PCR and direct sequence analysis in both directions using the ABI Big Dye Terminator Cycle Sequencing Ready Reaction kit and the ABI3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). This analysis also included the recently identified fourth exon by Bedard et al.25 (ENST00000370606). The Mutation Surveyor DNA Variant Analysis Software (SoftGenetics, State College, PA, USA) was used for the DNA sequencing analysis and variant calling. Full details of primer sequences, amplicon lengths and PCR conditions can be obtained upon request. Sequences were aligned with known ZIC3 coding reference sequence. Numbering of variants started at the A of the ATG translation initiation codon using Genbank mRNA reference sequence NM_003413.3 and genomic reference sequence NG_008115.1 according to the Human Genome Variation Society (HGVS) nomenclature.26 Variants were confirmed on DNA extracted from the second blood sample if available.
The pathogenicity of the variants was based on a number of genetic criteria: predicted non-functionality of mutated proteins; location; type and conservation of mutated amino-acids; segregation of clinical phenotypes in mutation carriers; presence/absence in control populations; and functional tests performed in this study. In silico prediction of functional consequences was performed using Align GVGD (http://agvgd.iarc.fr/agvgd_input.php), SIFT (http://blocks.fhcrc.org/sift/SIFT.html), PolyPhen (http://genetics.bwh.harvard.edu/pph) and MutationTaster (http://www.mutationtaster.org). Effects on splicing, if applicable, were investigated in silico using http://www.fruitfly.org/seq_tools/splice.html, http://www.cbs.dtu.dk/services/NetGene2, http://www.genet.sickkids.on.ca/~ali/splicesitefinder.html and http://rulai.cshl.edu/tools/ESE. The final classification of the detected variants was made on the basis of the classification system proposed by Plon et al.27 All variants detected in this study as well as all other variants described in literature previously were submitted to the LOVD database (http://www.LOVD.nl/ZIC3), with patient IDs ZIC3_000001 to ZIC3_000039.
Expression plasmid construction and mutagenesis
Total RNA was obtained from the human heart or brain total RNA kit (Clontech Laboratories, Inc, Mountain View, CA, USA) and complementary DNA was synthesized using the Quanta qScript cDNA Supermix (Quanta Biosciences, Beverly, MA, USA) as indicated by the manufacturer. The full-length ZIC3 cDNA was obtained using primers ZIC3F-5′-ATGACGATGCTCCTGGACGGAGGC-3′ and ZIC3R-5′- GGTTTGTGTTTGTCCTCAGACGTACCA-3′ and then ligated into the pGEM-Teasy vector using T4 DNA ligase (Promega, Madison, WI, USA). The ZIC3 variants c.680dup, c.842_843del and c.1111A>C were introduced with the Quick-Change II XL Site-Directed Mutagenesis (SDM) kit according to the manufacturer’s instructions (Stratagene, La Jolla, CA, USA). Details on SDM primer sequences can be obtained from the authors upon request. Wild-type (WT) and mutated ZIC3 pGEM-Teasy constructs were digested with EcoR1 and subcloned into pEGFPc1 and pCS2+ expression-vectors. All constructs, correct orientation of inserts as well as the presence of specified variants were verified by Sanger sequencing before further investigations.
Cell culture, transfection and subcellular localization
HeLa cells were grown in DMEM (Dulbecco’s Modified Eagle Medium, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS heat inactivated, PAA, A15-104) and 1 × Pen/Strep (Thermo Fisher Scientific) maintained at 37 °C and 5% CO2. Cells were detached using trypsin and washed with PBS pbefore further analysis. HeLa cells were seeded 16 h before transfection in 12-well plates on circular glass coverslips in DMEM complete medium. Plasmids were transfected using the Fugene 6 Transfection Kit (Promega) according to the manufacturer’s instructions. After 48 h, the transfected cells were washed in PBS and fixed for 10 min at room temperature in 4% paraformaldehyde/PBS. Following two washes in PBS, slides were mounted in Vectashield medium H-1200 (Vector Laboratories, Burlingame, CA, USA) containing 4’,6 diamidino-2-phenylindole (DAPI) to visualize nuclei. Cells were scored in three different categories regarding ZIC3 protein localization, respectively 1: cytoplasmic, 2: nuclear and cytoplasmic and 3: nuclear. A minimum of 50 cells across two independent experiments were scored for each ZIC3 construct.
Fish lines, mRNA synthesis, injections and phenotypic analysis
The following zebrafish lines were used: Tg(myl7:GFP)28 and TL. Fish were maintained according to standard laboratory conditions.29 Animal experiments were approved by the Animal Experimentation Committee (DEC) of the Royal Netherlands Academy of Arts and Sciences. Synthetic capped mRNA was generated by transcribing 1 μg of the appropriate plasmids using the SP6 mMessage mMachine kit (Thermo Fisher Scientific). Subsequently, 100 pg of mRNA was injected into 1-cell stage zebrafish embryos. Embryos were examined morphologically at 28 h post fertilization (hpf) and scored for heart position and other (midline) defects. For each construct a minimum of 80 embryos were injected and scored. The scoring system was divided in five categories respectively (1) lethal, (2) severe posterior truncation, (3) right-sided heart, (4) midline heart and (5) normal left-sided heart.
Results
Identification of ZIC3 variants
In total 348 unrelated individuals were tested for mutations in the ZIC3 gene. Congenital heart disease was very variable in the patient group (Table 1). Out of the 348 patients ~33% had heterotaxy (classic, CHD, abdominal or situs inversus), 26% had complex CHD, 16% cyanotic heart disease, 16% septation defects and 9% other. There was a predominance in male versus female patients (55% vs 39%) and the population was very young (66% diagnosed <1 year).
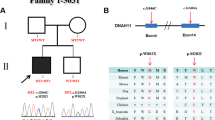
Six probably pathogenic variants were identified in six unrelated families, of which three were frame-shift variants, one nonsense variant, one missense variant and one splice-site variant (Supplementary Table 1). Whenever possible both symptomatic and asymptomatic family members of index patients were tested for the identified variants. Representative pedigrees and tested family members are depicted in Figure 1. For five of the six described families, DNA was available from a least one member with classic heterotaxy. For family 23 081 we could only test the asymptomatic mother, because DNA of her (affected) deceased son and two deceased brothers was not available. The index patient of family 18 173 is a male neonate with heterotaxy and caudal regression syndrome. Two prior male pregnancies were terminated due to similar findings. The index was found to have a hemizygous variant c.680dup, predicting a frame-shift and premature stop codon p.(Val228Serfs*50). In one of the affected fetuses, the mother and the grandmother the variant was also present. Index patients of family 19 018 were two male fetuses that were aborted at 22 weeks pregnancy due to prenatal detection of heterotaxy. Both were hemizygous for the c.842_843del variant also predicting a frame-shift and premature stop codon p.(His281Argfs*62). The mother was heterozygous carrier of the variant, while the grandmother lacked the variant. The grandfather was not available for testing. The index of family 23 217 was a second terminated male pregnancy with Ivemark syndrome (see Figure 2a and b). This fetus was hemizygous for variant c.869del, predicting a frame-shift and aberrant translated protein p.(Pro290Argfs*118). No DNA was available of other family members. The index of family 27 983 was a male fetus with a complex cor vitium (DORV, TGA and AVSD) and isomerism. The pregnancy was terminated at 21 weeks. He was found to have a hemizygous splice-site variant c.1060+1G>A. His asymptomatic mother and grandmother are both heterozygous for the variant. Index of family 23 081 is an asymptomatic female of whom a son and two brothers died neonatally. The deceased son had situs inversus, AVSD, PS and malposition of the great arteries and died soon after birth. The two brothers died at age 5 months. They both had situs inversus, anus atresia and a complex heart defect. She was found to carry a nonsense variant c.1063G>T predicting a premature stop codon p.(Glu355*). No DNA of the male members had been preserved for DNA testing. Two of her daughters with a desire to have children did not have the variant. In family 21 753 the first male fetus with malformations was discovered with ultrasound at 19 weeks. The pregnancy was terminated at 21 weeks due to serious cardiac abnormalities. Autopsy revealed truncus arteriosus type I, large inlet VSD, trilobar lungs, preauricular tag and clump feet. The pregnancy of the second male fetus, the index patient, was terminated at 15 weeks+6 days due to suspected TGA. Autopsy revealed complex CHD with TGA, dextrocardia, AVSD and abnormal pulmonal and systemic venous return. Non-cardiac abnormalities included asplenia and a symmetrical liver with left-sided galbladder. Fully detailed autopsy photographs are depicted in Figure 2c and h. In both fetuses variant c.1111A>C p.(Asn371His) was identified which is located in the fourth zinc-finger domain. The mother as well as her sister were heterozygous for the variant. Her asymptomatic father was hemizygous carrier of the variant.


Overview of variants in the ZIC3 gene. (a) Pedigrees of ZIC3 variant positive families. Squares, males; circles, females; diamonds with 'm', male stillborns/terminations; +, hemizygous mutation; +/−, heterozygous mutation; −/−, wild-type. Arrows indicate indexes. Reference sequences are based on Genbank accession numbers NM_003413.3, NP_003404.1 and NG_008115.1. Below pedigrees are representative Sanger sequences of the variants with the upper line being the reference sequence, the middle line a hemizygous male carrier and the bottom line a heterozygous female carrier. (b) Representation of the ZIC3 gene (NM_003413.3) and corresponding ZIC3 protein (NP_003404.1). On the right all disease associated variations known in literature. Black: pathogenic variants; blue: VUS. On the left variations detected in this study, in red, probably pathogenic mutations; blue, VUS. NES, nuclear export signal; NLS, nuclear localization signal; VUS, variants of unknown significance.

(a,b) Autopsy report photographs of the male index fetus of family 23 217. (a) Opened thorax and abdomen with situs inversus. (b) Graphic representation of situs inversus. (c–h) Autopsy report photographs of male index fetus of family 21 753 (c) heart from the back, (d) heart from the left side, (e) opened left ventricle, (f) organs seen from the backside, (g) opened thorax and abdomen and (h) liver from the backside. A detailed clinical description of the fetus is described in the result section.
Five other index patients were found to have a variation in the N-terminal poly-alanine repeat region. Three patients had a 10–11 expansion; one male neonate with TGA and obstructive foramen ovale. His mother also carried the variant; one male patient with situs inversus and one female patient with type II ASD. This 10–11 expansion was also detected in 4/336 in-house control individuals. Two patients had a 10 to 7 poly-alanine contraction due to a deletion of nine base pairs. One was a male with VSD and ASD type II. His mother and her asymptomatic brother also had the variant. The second patient was a female patient with situs inversus, dextrocardia and VSD. This variant was not detected in 336 control individuals.
Altered subcellular localization of mutated ZIC3 proteins
The subcellular distribution of mutated ZIC3 proteins was assessed as one of the potential factors contributing to protein dysfunction, when not localized in the nucleus. Transient transfections in HeLa cells of GFP-tagged wild-type (WT) and mutated ZIC3 proteins were performed to determine the fractions of nuclear/cytoplasmic situated proteins. At least 50 cells per construct were evaluated blindly by two expert laboratory technicians and ZIC3 localization was scored in three categories, as described in the materials and methods section. Both the WT and missense variant p.(Asn371His) localized exclusively in the nucleus of all cells. Truncated ZIC3 proteins localized both in the nucleus and the cytoplasm, albeit for mutated protein p.(His281Argfs*62) more in the nucleus than the cytoplasm, whilst mutated protein p.(Val228Serfs*50) was equally distributed over nucleus and cytoplasm. Representative localization patterns are visualized in Figure 3a–d and division over the three categories is summarized in Figure 3e.

Representative subcellular localization of WT and mutated ZIC3 proteins. (a–d) First column represent WT or mutant GFP-tagged ZIC3 proteins transfected in HeLa cells. Second column is the nuclear DAPI staining and the third column is both columns merged. A minimum of 50 cells were scored for each construct. Scoring was based on three localization classifications: nuclear, cytoplasmic or both (e).
Overexpression of mutant ZIC3 proteins in zebrafish
Zebrafish are an elegant model to monitor the effect of transcription factor mutations on biological activity in vivo, as other studies have shown previously.30 At 28 h post fertilization the linear heart tube is formed in zebrafish embryos and is positioned under the left eye (Figure 4a). Injection of 100 pg mRNA encoding human WT ZIC3 in one-cell stage fish embryo’s resulted in >40% of embryos with a heart tube positioned either at the midline or under the right eye indicative for a defect in left-right axis formation (Figure 4). Besides these cardiac laterality defects injection of WT ZIC3 mRNA also induced more severe phenotypes such as posterior truncation or embryo lethality. Injections of equal amounts mRNA encoding for the p.(His281Argfs*62) or the p.Val288Serfs*50 variants resulted in predominantly WT embryos demonstrating that these are loss-of-function alleles. Injection of mRNA encoding for the p.(Asn371His) variant induced cardiac laterality defects or more severe phenotypes, however, to a lesser extent compared with injection of WT mRNA, suggesting a partial loss of function. A minimum of 80 embryos was analyzed for each construct.

Functional assessment of variants in zebrafish. (a) Representative micrographs of cmlc2:GFP zebrafish embryo’s displaying cardiac laterality defects. The picture on the left shows a normally located left-sided heart (left-sided heart), while the picture in the middle indicates an intermediately positioned heart (midline heart). The picture on the right illustrates an aberrantly located right-sided heart (right-sided heart). (b) Quantification of phenotypes upon expression of wild-type human ZIC3 protein and of the indicated variants. A minimum of 80 embryos was analyzed. uic, uninjected control.
Discussion
In current literature 33 distinct pathogenic variants and several variants of unknown significance (VUS) have been described in the ZIC3 gene in heterotaxy patients (Figure 1b and Supplementary Table 1), providing substantial evidence that loss-of-function or aberrant functioning of the ZIC3 protein during embryogenesis is a cause of heterotaxy. In this study, we screened 348 patients for the ZIC3 gene. These patients were referred to our Department of Clinical Genetics for diagnostic testing due to various cardiac malformations (Table 1) over a time period>10 years. In agreement with previous reports variants were detected in both males and females. The class 5 variants (pathogenic) were detected only families with a clear or suggestive clinical history indicative for X-linked inheritance. No pathogenic variants were identified in sporadic patients. In family 19 018, the maternal grandmother was proven not to be carrier of the variant (Figure 1a), therefore the variant is either de novo in the mother or present in the asymptomatic grandfather. A definite genotype diagnosis was seen in family 21 753 were the maternal grandfather proved to be asymptomatic carrier of the familial variant. This incomplete penetrance in a male carrier has been reported twice before, where one possible explanation was ascribed either to a residual portion of functional protein expression or compensation by other redundant proteins.31, 32 One variant in these publications was p.(Lys408*), located at the very near end of the last zinc finger. As all major important domains are all still present this possibly explains residual activity. Our in vivo experiments of the detected mutation in this family, in which partial loss of function was seen, may confirm this hypothesis. The second variant p.Arg350Gly is a very conserved aminoacid in ZF3. The family is this family had 7 affected males and one unaffected. It remains unclear why this one male was unaffected. As stated, we did not detect any pathogenic or likely pathogenic variants in sporadic patients similar to previous reports.31, 33, 34 Additionally we did not identify any suspicious variants in patients with isolated TGA (30 patients with TGA, included in the subgroup cyanotic heart disease) comparable to earlier reports.24, 31, 34
The final classification of variants in this study was based on a combination of in silico analysis as described in the Materials and Methods, together with functional data generated in this study as well as data from earlier publications. A summary of these data is presented in Supplementary Table 1 and Figure 1b. Six variants were determined to be causative, of which five were presumably truncating variants and one was a missense variant (Figure 1 and Supplementary Table 1). The truncating variants included one nonsense variant p.(Glu355*), three frame-shift variants p.(Val228Serfs*50), p.(His281Argfs*62), p.(Pro290Argfs*118) and one splice-site variant (c.1060+1G>A). All these variants are located either upstream or within one of the zinc-finger domains, thereby altering important regions of the ZIC3 protein. None of these variants have been reported before and are absent from all relevant databases. This study is the first to report on a splice-site variant in the ZIC3 gene responsible for heterotaxy, indicating that all types of variants can occur in X-linked heterotaxy families and that most pathogenic variants are private. The one missense variant identified p.(Asn371His) in this study is located in the fourth zinc-finger domain and is expected to be damaging by all four predictive software programs. We also detected several variations in the N-terminal alanine-tract (Figure 1b and Supplementary Table 1) as seen in other studies.34, 35 We detected the known single Ala expansion (10–11) three times. This single expansion Ala was also detected in in-house controls, in both male and female individuals. Together with the functional data of Cowan et al.,34 in which transactivation activity and nuclear localization are unaltered, it seems that this single expansion is at most a risk factor, but not solely causative. In addition, we also detected a contraction of the Ala repeat with three residues (10 to 7) in two patients. However not detected in controls, this contraction was also found in at least one asymptomatic male carrier, also contradicting a clear pathogenic character of this region. However, since Wessels et al.35 detected a de novo expansion of 10–12 Ala residues and not all these variants have been functionally tested, the relevance of these variants remain to be determined. When assessing nuclear localization of mutated ZIC3 protein the two frame-shift variants p.(Val228Serfs*50) and p.(His281Argfs*62) are predicted to disrupt the previously mapped nuclear localization and export signals (NLS/NES)36, 37 and localization of these truncated proteins was divided over both nucleus and cytoplasm (Figure 2b and c) in line with Cowan et al.34 However, mutated p.(Asn371His) ZIC protein, predicted to disrupt one of the mapped NLS and located in a very conserved region in the ZF4 domain showed no different cell localization compared with WT protein and was completely localized to the nucleus in our study (Figure 3a and d). Bedard et al. 36 showed that fragments in this region (350–380, 340–420 and 340–466) showed a mixture of cytoplasmic and nuclear localization and that a mutation in one of these regions might be sufficient to disrupt nuclear localization. Our study shows that at least the majority of p.(Asn371His) mutated protein is located in the nucleus. Zebrafish are an excellent model to study aberrant gene function during development, specifically genes involved in the cardiovascular system.38 During the first days of development zebrafish embryos are not dependent on a functional cardiovascular system and as such embryos with severe cardiovascular defects can be studied. Zic3 expression is restricted to the dorsal site during gastrulation before laterality is established.39 Overexpression of wild-type Zic3 (by the mRNA injection) will disturb this pattern and result in ectopic Zic3 activity (eg, The ventral side of the embryo). This explains the posterior truncations seen in the embryos injected with the wt zic3 mRNA (Figure 4b). In this way the ZIC3 mutant protein were assessed by analyzing the functional defects after overexpression. Clearly, both frameshift variants p.(Val228Serfs*50) and p.(His281Argfs*62) that predict an early truncation of the ZIC3 protein before the zinc fingers showed non-functionality as the results resemble the uninjected controls (uic, Figure 4). So either these aberrant proteins have no function or (part of) the mRNA transcripts are eliminated via nonsense-mediated mRNA decay (NMD). On the contrary the p.(Asn371His) ZIC3 protein partly retains functionality, which is supported by its nuclear localization. The mechanism by which this mutated protein still leads to heterotaxy therefore is likely due to decreased ZIC3 protein expression and/or timely activity. Concluding, in a cohort of 348 patients with a wide range of congenital heart disease we have identified six novel pathogenic ZIC3 variants in heterotaxy families. If the patient cohort is split into 'heterotaxy' and 'non-heterotaxy' patients the frequency of ZIC3 mutations is relatively high in the former group (6/114=5,3%) and absent in the latter group (0/234=0%).
In addition, we are the first to assess the functional consequences of three ZIC3 variants in both cell lines and a zebrafish model indicating varying degrees of loss-of-function consequences of mutated proteins. The combination of results in this study expands the knowledge on type, effect and clinical implications of ZIC3 variants and will help in counseling affected families.
References
Hoffman JI, Kaplan S : The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39: 1890–1900.
van der Linde D, Konings EE, Slager MA et al: Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol 2011; 58: 2241–2247.
Epstein JA : Franklin H. Epstein Lecture. Cardiac development and implications for heart disease. N Engl J Med 2010; 363: 1638–1647.
Fahed AC, Gelb BD, Seidman JG, Seidman CE : Genetics of congenital heart disease: the glass half empty. Circ Res 2013; 112: 707–720.
Shiraishi I, Ichikawa H : Human heterotaxy syndrome - from molecular genetics to clinical features, management, and prognosis. Circ J 2012; 76: 2066–2075.
Francis RJ, Christopher A, Devine WA, Ostrowski L, Lo C : Congenital heart disease and the specification of left-right asymmetry. Am J Physiol Heart Circ Physiol 2012; 302: H2102–H2111.
Bruneau BG : The developmental genetics of congenital heart disease. Nature 2008; 451: 943–948.
Sutherland MJ, Ware SM : Disorders of left-right asymmetry: heterotaxy and situs inversus. Am J Med Genet C Semin Med Genet 2009; 151C: 307–317.
Vitale E, Brancolini V, De Rienzo A et al: Suggestive linkage of situs inversus and other left-right axis anomalies to chromosome 6p. J Med Genet 2001; 38: 182–185.
Casey B, Cuneo BF, Vitali C et al: Autosomal dominant transmission of familial laterality defects. Am J Med Genet 1996; 61: 325–328.
Eronen M, Kajantie E, Boldt T, Pitkanen O, Aittomaki K : Right atrial isomerism in four siblings. Pediatr Cardiol 2004; 25: 141–144.
Casey B, Devoto M, Jones KL, Ballabio A : Mapping a gene for familial situs abnormalities to human chromosome Xq24-q27.1. Nat Genet 1993; 5: 403–407.
Gebbia M, Ferrero GB, Pilia G et al: X-linked situs abnormalities result from mutations in ZIC3. Nat Genet 1997; 17: 305–308.
Mohapatra B, Casey B, Li H et al: Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet 2009; 18: 861–871.
Goldmuntz E, Bamford R, Karkera JD, dela Cruz J, Roessler E, Muenke M : CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am J Hum Genet 2002; 70: 776–780.
Ma L, Selamet Tierney ES, Lee T, Lanzano P, Chung WK : Mutations in ZIC3 and ACVR2B are a common cause of heterotaxy and associated cardiovascular anomalies. Cardiol Young 2012; 22: 194–201.
Kaasinen E, Aittomaki K, Eronen M et al: Recessively inherited right atrial isomerism caused by mutations in growth/differentiation factor 1 (GDF1). Hum Mol Genet 2010; 19: 2747–2753.
Tariq M, Belmont JW, Lalani S, Smolarek T, Ware SM : SHROOM3 is a novel candidate for heterotaxy identified by whole exome sequencing. Genome Biol 2011; 12: R91.
Perles Z, Cinnamon Y, Ta-Shma A et al: A human laterality disorder associated with recessive CCDC11 mutation. J Med Genet 2012; 49: 386–390.
Houtmeyers R, Souopgui J, Tejpar S, Arkell R : The ZIC gene family encodes multi-functional proteins essential for patterning and morphogenesis. Cell Mol Life Sci 2013; 70: 3791–3811.
Paulussen AD, Schrander-Stumpel CT, Tserpelis DC et al: The unfolding clinical spectrum of holoprosencephaly due to mutations in SHH, ZIC2, SIX3 and TGIF genes. Eur J Hum Genet 2010; 18: 999–1005.
Purandare SM, Ware SM, Kwan KM et al: A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development 2002; 129: 2293–2302.
Ware SM, Harutyunyan KG, Belmont JW : Zic3 is critical for early embryonic patterning during gastrulation. Dev Dyn 2006; 235: 776–785.
D'Alessandro LC, Latney BC, Paluru PC, Goldmuntz E : The phenotypic spectrum of ZIC3 mutations includes isolated d-transposition of the great arteries and double outlet right ventricle. Am J Med Genet A 2013; 161A: 792–802.
Bedard JE, Haaning AM, Ware SM : Identification of a novel ZIC3 isoform and mutation screening in patients with heterotaxy and congenital heart disease. PLoS One 2011; 6: e23755.
den Dunnen JT, Antonarakis SE Mutation nomenclature; in Jonathan L Haines et al (eds). Current Protocols in Human Genetics/Editorial Board. 2003, Chapter 7, Unit 7 13.
Plon SE, Eccles DM, Easton D et al: Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 29: 1282–1291.
Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ : Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn 2003; 228: 30–40.
Lawrence C : The husbandry of zebrafish (Danio rerio): A review. Aquaculture 2007; 269: 1–20.
Domene S, Roessler E, El-Jaick KB et al: Mutations in the human SIX3 gene in holoprosencephaly are loss of function. Hum Mol Genet 2008; 17: 3919–3928.
Megarbane A, Salem N, Stephan E et al: X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. Eur J Hum Genet 2000; 8: 704–708.
D'Alessandro LC, Casey B, Siu VM : Situs inversus totalis and a novel ZIC3 mutation in a family with X-linked heterotaxy. Congenit Heart Dis 2013; 8: E36–E40.
Ware SM, Peng J, Zhu L et al: Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet 2004; 74: 93–105.
Cowan J, Tariq M, Ware SM : Genetic and functional analyses of ZIC3 variants in congenital heart disease. Hum Mutat 2014; 35: 66–75.
Wessels MW, Kuchinka B, Heydanus R et al: Polyalanine expansion in the ZIC3 gene leading to X-linked heterotaxy with VACTERL association: a new polyalanine disorder? J Med Genet 2010; 47: 351–355.
Bedard JE, Purnell JD, Ware SM : Nuclear import and export signals are essential for proper cellular trafficking and function of ZIC3. Hum Mol Genet 2007; 16: 187–198.
Hatayama M, Tomizawa T, Sakai-Kato K et al: Functional and structural basis of the nuclear localization signal in the ZIC3 zinc finger domain. Hum Mol Genet 2008; 17: 3459–3473.
Bakkers J : Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res 2011; 91: 279–288.
Grinblat Y, Sive H : zic Gene expression marks anteroposterior pattern in the presumptive neurectoderm of the zebrafish gastrula. Dev Dyn 2001; 222: 688–693.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Paulussen, A., Steyls, A., Vanoevelen, J. et al. Rare novel variants in the ZIC3 gene cause X-linked heterotaxy. Eur J Hum Genet 24, 1783–1791 (2016). https://doi.org/10.1038/ejhg.2016.91
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.91
This article is cited by
-
Whole exome sequencing of fetal structural anomalies detected by ultrasonography
Journal of Human Genetics (2021)
-
A novel ZIC3 gene mutation identified in patients with heterotaxy and congenital heart disease
Scientific Reports (2018)
