
Abstract
Germline variants in subunit D of the succinate dehydrogenase gene (SDHD variants) are associated with an increased risk of developing paragangliomas. The aim of this study was to compare mortality rates and survival in a Dutch cohort of SDHD variant carriers with those in the general population. The study was conducted at the Leiden University Medical Center, a tertiary referral center for patients with paragangliomas. Included subjects all tested positive for SDHD variants before 1 July 2012 and visited the departments of Otorhinolaryngology or Endocrinology at least once or had a diagnosed paraganglioma and a SDHD variant-positive family history. Clinical data were retrieved from medical records, information on mortality was obtained from the Municipal Personal Records Database, and mortality rates for the Dutch population were obtained from the Dutch Central Bureau of Statistics, stratified by sex, age and date. SDHD variant carriers were followed from the date of first SDHD variant-related contact until death, emigration or 12 December 2012 and the standardized mortality ratio (SMR) was calculated. Two-hundred and seventy-five SDHD variant carriers were included in the study, of which 80% carried the c.274G>T, p.(Asp92Tyr) variant, had a mean duration of follow-up of 7.6 years, yielding 2242 person-years of observation for analysis. There were 18 deaths in the SDHD variant carrier group; two were paraganglioma related. The SMR for the whole cohort was 1.07 (95% confidence interval 0.67–1.73). In conclusion, mortality in SDHD variant carriers is not substantially increased. Additional studies are required to confirm these findings.
Similar content being viewed by others

Introduction
Germline variants in subunit D of the succinate dehydrogenase (SDH) gene predispose carriers to the development of paragangliomas (PGLs).1 SDHD variants are mainly associated with multifocal PGLs in the head and neck region (HNPGLs), although sympathetic PGLs (sPGLs; extra-adrenal PGLs) and adrenal PGLs (ie, pheochromocytomas, PCC) also occur.2, 3, 4 Although the majority of HNPGLs are benign and indolent tumors,5 their location in close proximity to important neurovascular structures may lead to serious morbidity.6 Neurovascular complications occur in up to 60% of cases following surgical treatment, for example, cranial nerve injury and lesions to the carotid artery.7, 8 It is therefore of great importance to carefully consider whether HNPGL should be treated, and a ‘wait and scan’ policy is often the best option.5
Because of their ability to hypersecrete catecholamines, PCC and sPGLs can give rise to severe cardiovascular complications, such as shock, myocardial infarction, dissecting aortic aneurysms or heart failure due to toxic cardiomyopathy.9, 10, 11, 12 In order to avoid these potentially lethal complications, adrenalectomy is indicated for PCC,13 and with implementation of appropriate preoperative care to modulate the effects of catecholamine release, perioperative mortality is nil.14, 15, 16
The pooled incidence of malignant PGL, defined as the presence of metastases,17, 18, 19 in populations comprising both unaffected SDHD variant carriers and SDHD variant carriers with manifest non-malignant PGL is about 8%.20 Prognosis in malignant PGL is poor, with reported 5-year survival rates of 20–55% for malignant sPGL and PCC21, 22 and 60% for malignant HNPGL,23 although a few cases of survival for >20 years after diagnosis have been described.24, 25
An increasing number of SDHD variant carriers are now being identified through (presymptomatic) testing of family members of SDHD variant carriers with manifest disease, that is, index cases. It is important that these newly identified variant carriers receive reliable prognostic information, including the impact of SDHD variants on mortality and survival. As SDHD variants are associated with a high risk for HNPGLs, and fatal cases of (untreated) PCC have been described, it is important to know whether this translates into increased mortality risk. As this question has not yet been addressed for SDHD variant carriers, the objective of this study was to compare mortality rates and survival in a Dutch cohort of SDHD variant carriers with that of the general Dutch population.
Subjects and methods
Eligibility criteria
The database of the Laboratory for Diagnostic Genome Analysis of the Leiden University Medical Center (LUMC), a tertiary referral center for patients with PGLs, was used to identify carriers of SDHD variants. Screening for SDH variants was performed in all persons diagnosed with PGL who agreed to genetic testing. Informed consent of both parents was required for individuals aged between 12 and 16 years.
In index patients, all exonic and adjacent intronic regions of the SDH genes were tested for the presence of variants by direct sequencing using the Sanger method on an ABI 377 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA) and multiplex ligation-dependent probe amplification (MLPA) was carried out with the P226 MLPA Kit (MRC Holland, Amsterdam, the Netherlands).3 Family members of index patients were tested for the family-specific variant. The reference sequence NG_012337.1 covering SDHD transcript NM_003002.2, available from the TCA Cycle Gene Variant Database LOVD database, was used to describe variants. All variants described in this study were previously submitted to the above LOVD database (http://chromium.liacs.nl/lovd_sdh).
Consecutive SDHD variant carriers who tested positive before 1 July 2012 and who visited the departments of Otorhinolaryngology or Endocrinology at least once were included. In addition, persons with a PGL diagnosis and a SDHD variant-positive family history and known to the outpatient clinics of the departments of Endocrinology and Otorhinolaryngology of the LUMC were also included, as they were considered to be obligate SDHD variant carriers. Only SDHD variant carriers with paternal inheritance were included. Persons for whom no information could be retrieved at the Municipal Personal Records Database (see below) were excluded.
Clinical characteristics
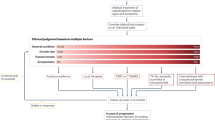
Clinical data were retrieved from medical records. Since 2002, a standard evaluation protocol has been implemented at the departments of Endocrinology and Otorhinolaryngology (Figure 1). In order to detect (hormonally active) PGLs, biochemical screening and head-and-neck magnetic resonance imaging (MRI) were performed at intervals of 2 years (at 3-yearly intervals in unaffected variant carriers). The original diagnostic protocol for patients in the period before 2002 was identical to that from 2002 onwards, with the exception of protocolized follow-up every 2 years.

Screening policy in SDHD variant carriers.
Biochemical screening included the measurement of (nor)epinephrine, vanillylmandelic acid and dopamine in two 24-h urinary samples. From 2005 onwards, (nor)metanephrine and 3-methoxytyramine were added to these measurements.4 In cases with excessive catecholamine secretion (ie, any value above the upper reference limit), radiological assessment by MRI or computed tomographic scans of thorax, abdomen and pelvis was performed to identify potential sources of excessive catecholamine production outside the head and neck region, followed by whole-body 123I metaiodobenzylguanidine scans if a suspected lesion was found. Patients with PCC or sPGLs operated at the LUMC are generally prepared with adequate preoperative α- and, if necessary, β-adrenergic blockade. In all surgically resected PGLs, diagnosis was confirmed by pathological investigation.
Mortality and survival
For this study, follow-up data on SDHD variant carriers were included from the date of genetic testing. In cases where clinical follow-up was available for the period before SDHD genetic testing, this period was not considered in the mortality analysis, because it would have introduced immortal time bias with an underestimation of mortality rates.26 Because our aim was to determine the relation between mortality and carriage of a SDHD variant and not between mortality and a diagnosis of PGL, obligate SDHD variant carriers were included from their first PGL-related contact at the LUMC. We also included these patients in the mortality analyses.
Follow-up ended 12 December 2012, or at date of death or, in case of emigration, on the date of emigration. Ten patients are currently being followed-up at another hospital.
For all included (obligate) SDHD variant carriers, an enquiry was sent to the Municipal Personal Records Database (GBA) on 12 December 2012. The GBA registers all deaths of Dutch inhabitants. To compare mortality between (obligate) SDHD variant carriers and the general population, the standardized mortality ratio (SMR) was estimated. Mortality rates for the Dutch population were obtained from the Dutch Central Bureau of Statistics (The Netherlands), using rates stratified by sex, age (per 1 year) and date (1-year periods). The SMR was calculated by dividing the observed number of deaths in the SDHD cohort, and the expected number of deaths calculated as the sum of the stratified number of expected deaths (stratum specific mortality rates from the general population times follow-up time at risk). Post-hoc power calculations revealed sufficient power (>0.8) to detect a difference in mortality of >7%.
Survival curves were produced for SDHD variant carriers and the general population, depicting observed and expected survival, respectively. STATA 12.0 (Stata Corp, College Station, TX, USA) was used for statistical analysis.
Results
Clinical characteristics
Of the 275 SDHD variant carriers included, 131 (48%) were female. Clinical characteristics are detailed in Table 1. Molecular genetic testing was used to identify 193 cases (70%), with the remaining 82 individuals characterized as obligate SDHD variant carriers. The SDHD c.274G>T, p.(Asp92Tyr) variant (SDHD LOVD ID: SDHD_00004) was present in 80% of the cohort, 11% carried the SDHD c.416T>C, p.(Leu139Pro) variant (SDHD LOVD ID: SDHD_00016) and 3% the SDHD c.284T>C, p.(Leu95Pro) variant (SDHD LOVD ID: SDHD_00039). Six other SDHD variants were found in the remaining subjects.
Mean age at first identification of a SDHD variant or PGL-related contact at the LUMC was 43.5±14.4 years. By the end of follow-up, a total of 620 HNPGLs had been found in 246 patients (89%), with 200 patients showing multiple HNPGLs. Treatment was initiated for 143 patients (52%), and a ‘wait and scan’ policy was chosen for the remaining cases. The primary therapy option was surgery, and only seven patients were exclusively treated with radiotherapy or embolization. The disease status of five patients (2%) could not be determined, because these patients declined radiological imaging of the head and neck region owing to an absence of the symptoms.
In total, 231 (84%) patients underwent biochemical screening at least once at the department of Endocrinology, thereby identifying 20 sPGLs and 18 PCCs. The remaining 44 patients were seen at the department of Otorhinolaryngology. A ‘watchful waiting’ policy was adopted for four sPGL cases: the lesions were surgically difficult to assess, and because of a low risk of malignant transformation and the lack of symptoms related to catecholamine excess, a decision to postpone surgery was preferred. One patient with a non-secretory pheochromocytoma declined surgery. Fifteen patients with sPGL and 17 patients with pheochromocytoma were surgically treated, and the resection of one sPGL is scheduled. A malignant PGL was diagnosed in 10 patients (4%), and the primary tumor was a HNPGL in 7 of these cases. Finally, 20 individuals (7%) showed no signs of manifest disease during follow-up, that is, they were unaffected SDHD variant carriers.
Mortality, SMR and survival
Mortality data were available for all 275 cases, with none lost to follow-up. Over a mean follow-up period of 7.6 years (range 0–45 years), 18 SDHD variant carriers died at a mean age of 65 years. Of the 18 deaths in the SDHD variant carrier group, two were PGL related (ie, these people died of metastatic disease). Four people died of a cause not related to PGL, and of the 12 people, cause of death was unknown.
A total of 2242 person-years were available for comparison with normative data of the Dutch population. The SMR was 1.07 (95% confidence interval (CI) 0.67–1.73) for the whole cohort, indicating no substantial mortality in SDHD variant carriers compared with the general population. The SMR for female SDHD variant carriers was 1.24 (95% CI 0.56–2.76) and 1.00 (95% CI 0.55–1.81) for male carriers.
Survival curves of the SDHD variant carriers (‘observed survival’) and of the general population (‘expected survival’) are depicted in Figure 2.

Survival of SDHD variant carriers (observed survival) and of the general population (expected survival).
Discussion
The aim of the present study was to compare mortality rates and survival in SDHD variant carriers with those of the general population. Our results show that mortality in SDHD variant carriers is not substantially increased compared with the general population, despite the presence of HNPGL in a majority of variant carriers. As the prevalence of SDHD-related morbidity is high, these results convey an important message to (newly identified) SDHD variant carriers. However, this study cannot determine whether mortality risk and survival are influenced by the protocolized screening for catecholamine overproduction and subsequent surgical treatment of PCC/sPGL when necessary.
To the best of our knowledge, this is the first investigation of mortality and survival in SDHD variant carriers. The high prevalence of SDHD variants in the Netherlands and the long history of PGL research at the LUMC provided access to a large cohort of SDHD variant carriers with a long duration of follow-up. Although our results provided no evidence for substantially increased mortality in SDHD variant carriers, the upper limit of the confidence interval, at 1.73, indicates that some uncertainty remains. Therefore, additional large cohort studies are needed to confirm and expand these results.
The high prevalence of founder variants in the Netherlands is probably due to the fact that Dutch society was segregated based on socioeconomic and religious differences until the mid-twentieth century, leading to endogamy in isolated populations. This facilitated the proliferation of many Dutch founder variants.27 The c.274G>T, p.(Asp92Tyr) and c.416T>C, p.(Leu139Pro) founder variants in SDHD are the most prevalent cause of hereditary PGLs in the Netherlands;3, 4, 28 however, these specific variants are very rare in other series. Although this, strictly, prohibits simple generalization of our results to other cohorts of SDHD variant carriers, it is worth noting that no convincing genotype–phenotype correlation has ever been described for a specific disease-causing SDH variant.
Our main result—that survival in SDHD carriers is not substantially increased compared with the general population—may be surprising. Considering the poor prognosis of malignant PGL, an increased mortality in SDHD variant carriers would be expected if malignant PGL was common. However, the low incidence (8%) of malignant PGL in Dutch SDHD variant carriers probably means that it has little influence on overall mortality rates.4 An expected increase in mortality due to the serious morbidity that may result from (treatment of) HNPGLs and the potentially fatal course of (untreated) PCC6, 8, 29, 30 was not seen, which could be partly due to the LUMC follow-up policy. First, although 90% of SDHD variant carriers in our cohort developed a HNPGL, only 35% of these patients were treated. This ‘wait and scan’ policy may have resulted in a decreased treatment-related morbidity and possibly mortality. Second, the variant carriers included in our cohort were regularly screened for the presence of PCC. Early detection of PCC by screening results in a much lower prevalence of symptoms, lower catecholamine excess and smaller tumors compared with sporadic PCC detected by signs and symptoms.31 This may have led to a decrease in both disease- and treatment-related morbidity and, possibly, mortality. It should also be kept in mind that the present study may be underpowered to detect a small increase in mortality risk (ie, <7%).
Our center previously investigated the survival of Dutch patients diagnosed with a HNPGL between 1945 and 1960. In accordance with the present study, this earlier study was also unable to find a significant difference in survival compared with the general population.32 Although this earlier study did not assess variant status, as most HNPGL patients in the Netherlands are carriers of SDHD variants3 the majority of patients included in that study must have been SDHD variant carriers. This previous study reported results from an era before the regular screening of HNPGL patients for the presence of PCC. The prevalence of PCC in Dutch SDHD variant carriers is reported to be 9%,4 but even at this frequency no effect on mortality due to an absence of screening could be detected. In addition, the subjects included in this study are under regular medical surveillance, which may have resulted in the earlier detection of other clinical conditions and therefore lower than expected deaths compared with the general population.
Seven percent of the study cohort did not display any signs of manifest disease, that is, unaffected variant carriers. The number of unaffected variant carriers in our cohort is probably an underestimation of the actual number, as we were only able to include those under regular follow-up. Unaffected variant carriers are more likely to decline regular follow-up and may therefore remain under the clinical radar. Although this could have resulted in a more ‘diseased cohort’, a bias of this type implies that our findings on mortality are actually more robust.
In conclusion, mortality is not substantially increased in SDHD variant carriers. This knowledge brings a previously missing clarity to the prognostic outlook for (newly identified) variant carriers concerning the effect of SDHD variants on mortality and survival.
References
Baysal BE, Ferrell RE, Willett-Brozick JE et al: Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000; 287: 848–851.
Benn DE, Gimenez-Roqueplo AP, Reilly JR et al: Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91: 827–836.
Hensen EF, Siemers MD, Jansen JC et al: Variants in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clin Endocrinol (Oxf) 2011; 75: 650–655.
van Hulsteijn LT, den Dulk AC, Hes FJ, Bayley JP, Jansen JC, Corssmit EP : No difference in phenotype of the main Dutch SDHD founder variants. Clin Endocrinol (Oxf) 2013; 79: 824–831.
Jansen JC, van den Berg R, Kuiper A, van der Mey AG, Zwinderman AH, Cornelisse CJ : Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer 2000; 88: 2811–2816.
Papaspyrou K, Mann WJ, Amedee RG : Management of head and neck paragangliomas: review of 120 patients. Head Neck 2009; 31: 381–387.
Bradshaw JW, Jansen JC : Management of vagal paraganglioma: is operative resection really the best option? Surgery 2005; 137: 225–228.
Li-shan L, Chang-wei L, Heng G, Yue-hong Z, Xing-ming C, Yong-jun L : Efficacy of surgical therapy for carotid body tumors. Chin Med Sci J 2011; 26: 241–245.
Bergland BE : Pheochromocytoma presenting as shock. Am J Emerg Med 1989; 7: 44–48.
Park JH, Kim KS, Sul JY et al: Prevalence and patterns of left ventricular dysfunction in patients with pheochromocytoma. J Cardiovasc Ultrasound 2011; 19: 76–82.
Schurmeyer TH, Engeroff B, Dralle H, von zur Muhlen A : Cardiological effects of catecholamine-secreting tumours. Eur J Clin Invest 1997; 27: 189–195.
Triplett JC, Atuk NO : Dissecting aortic aneurysm associated with pheochromocytoma. South Med J 1975; 68: 748 753.
Pacak K, Eisenhofer G, Ahlman H et al: Pheochromocytoma: recommendations for clinical practice from the First International Symposium; October 2005: Nat Clin Pract Endocrinol Metab 2007; 3: 92–102.
Conzo G, Musella M, Corcione F et al: Laparoscopic adrenalectomy, a safe procedure for pheochromocytoma. A retrospective review of clinical series. Int J Surg 2013; 11: 152–156.
Kazic MR, Zivaljevic VR, Milan ZB, Paunovic IR : Perioperative risk factors, morbidity, and outcome of 145 patients during phaeochromocytoma resection. Acta Chir Belg 2011; 111: 223–227.
Kinney MA, Warner ME, vanHeerden JA et al: Perianesthetic risks and outcomes of pheochromocytoma and paraganglioma resection. Anesth Analg 2000; 91: 1118–1123.
Eisenhofer G, Bornstein SR, Brouwers FM et al: Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer 2004; 11: 423–436.
Linnoila RI, Keiser HR, Steinberg SM, Lack EE : Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol 1990; 21: 1168–1180.
Shapiro B, Sisson JC, Lloyd R, Nakajo M, Satterlee W, Beierwaltes WH : Malignant phaeochromocytoma: clinical, biochemical and scintigraphic characterization. Clin Endocrinol (Oxf) 1984; 20: 189–203.
van Hulsteijn LT, Dekkers OM, Hes FJ, Smit JW, Corssmit EP : Risk of malignant paraganglioma in SDHB-variant and SDHD-variant carriers: a systematic review and meta-analysis. J Med Genet 2012; 49: 768–776.
John H, Ziegler WH, Hauri D, Jaeger P : Pheochromocytomas: can malignant potential be predicted? Urology 1999; 53: 679–683.
Szalat A, Fraenkel M, Doviner V, Salmon A, Gross DJ : Malignant pheochromocytoma: predictive factors of malignancy and clinical course in 16 patients at a single tertiary medical center. Endocrine 2011; 39: 160–166.
Moskovic DJ, Smolarz JR, Stanley D et al: Malignant head and neck paragangliomas: is there an optimal treatment strategy? Head Neck Oncol 2010; 2: 23.
van den Broek PJ, de Graeff J : Prolonged survival in a patient with pulmonary metastases of a malignant pheochromocytoma. Neth J Med 1978; 21: 245–247.
Yoshida S, Hatori M, Noshiro T, Kimura N, Kokubun S : Twenty-six-years' survival with multiple bone metastasis of malignant pheochromocytoma. Arch Orthop Trauma Surg 2001; 121: 598–600.
Suissa S : Immortal time bias in pharmaco-epidemiology. Am J Epidemiol 2008; 167: 492–499.
Zeegers MP, van Poppel F, Vlietinck R, Spruijt L, Ostrer H : Founder variants among the Dutch. Eur J Hum Genet 2004; 12: 591–600.
Taschner PE, Jansen JC, Baysal BE et al: Nearly all hereditary paragangliomas in the Netherlands are caused by two founder variants in the SDHD gene. Genes Chromosomes Cancer 2001; 31: 274–281.
Kizer JR, Koniaris LS, Edelman JD, St John Sutton MG : Pheochromocytoma crisis, cardiomyopathy, and hemodynamic collapse. Chest 2000; 118: 1221–1223.
Newell KA, Prinz RA, Pickleman J et al: Pheochromocytoma multisystem crisis. A surgical emergency. Arch Surg 1988; 123: 956–959.
van Duinen N, Steenvoorden D, Bonsing BA et al: Pheochromocytomas detected by biochemical screening in predisposed subjects are associated with lower prevalence of clinical and biochemical manifestations and smaller tumors than pheochromocytomas detected by signs and symptoms. Eur J Endocrinol 2010; 163: 121–127.
de Flines J, Jansen J, Elders R et al: Normal life expectancy for paraganglioma patients: a 50-year-old cohort revisited. Skull Base 2011; 21: 385–388.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
van Hulsteijn, L., Heesterman, B., Jansen, J. et al. No evidence for increased mortality in SDHD variant carriers compared with the general population. Eur J Hum Genet 23, 1713–1716 (2015). https://doi.org/10.1038/ejhg.2015.36
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.36
This article is cited by
-
Feasibility and safety of the posterior retroperitoneoscopic approach in the resection of aortocaval and infrarenal paraganglioma: a single-center experience
Surgical Endoscopy (2021)
-
Clinical progression and metachronous paragangliomas in a large cohort of SDHD germline variant carriers
European Journal of Human Genetics (2018)
