
Abstract
Accurate estimation of cancer risk in HBOC families often requires BRCA1/2 testing, but this may be impossible in deceased family members. Previous, testing archival formalin-fixed, paraffin-embedded (FFPE) tissue for germline BRCA1/2 variants was unsuccessful, except for the Jewish founder mutations. A high-throughput method to systematically test for variants in all coding regions of BRCA1/2 in archival FFPE samples of non-tumor tissue is described, using HaloPlex target enrichment and next-generation sequencing. In a validation study, correct identification of variants or wild-type was possible in 25 out of 30 (83%) FFPE samples (age range 1–14 years), with a known variant status in BRCA1/2. No false positive was found. Unsuccessful identification was due to highly degraded DNA or presence of large intragenic deletions. In clinical use, a total of 201 FFPE samples (aged 0–43 years) were processed. Thirty-six samples were rejected because of highly degraded DNA or failed library preparation. Fifteen samples were investigated to search for a known variant. In the remaining 150 samples (aged 0–38 years), three variants known to affect function and one variant likely to affect function in BRCA1, six variants known to affect function and one variant likely to affect function in BRCA2, as well as four variants of unknown significance (VUS) in BRCA1 and three VUS in BRCA2 were discovered. It is now possible to test for germline BRCA1/2 variants in deceased persons, using archival FFPE samples from non-tumor tissue. Accurate genetic counseling is achievable in families where variant testing would otherwise be impossible.
Similar content being viewed by others


Introduction
There has been a tremendous technological development, since the discovery and cloning of the BRCA1 (breast cancer 1, early onset) gene in 19941 and shortly after the BRCA2 (breast cancer 2, early onset) gene in 1995.2 Today, testing for variants in the two genes is a widespread option, when counseling families with high risk of breast and ovarian cancer (HBOC). Women harboring a germline variant known to affect function in BRCA1 or BRCA2 are confronted with a lifetime risk of breast cancer of 60–80% and a risk of ovarian cancer of 20–50%3, 4 as well as risk of other cancers like pancreatic cancer and malignant melanoma.5 Men harboring the same variants are facing an increased risk of prostate cancer and breast cancer.5 Many variant carriers will choose prophylactic surgery to reduce their cancer risk or may enter more extensive screening programs, to detect cancer in an early stage to improve the outcome.6
Until now, testing for BRCA1 and BRCA2 variants (or germline variants in other high-risk genes), using traditional methods, required a blood sample, saliva sample or buccal smear from a living person or archived fresh frozen tissue or blood from a deceased person, in order to obtain high-quality DNA for the analysis. This has ruled out families, in which the relatives suffering from breast or ovarian cancer have already died. In such families, where, eg, a young woman is seeking genetic counseling, and her mother or other close relatives died from breast or ovarian cancer at a young age (eg, a decade ago), there will be no options for variant testing. If a variant testing is carried out in the woman seeking genetic counseling, a negative result will be difficult to interpret and cannot be used to predict her risk of breast and ovarian cancer. Today, the only option for such families is to use ‘indirect’ testing, where variant testing is offered to close relatives (siblings and children to the deceased person) to search for a germline variant.7 It is recommended to test at least 3–4 first-degree relatives, in order to increase the probability to identify or exclude a variant, which can make indirect testing costly and laborious. Furthermore, it can sometimes be difficult (or impossible) to get blood samples from relatives. Lastly, in many countries, indirect testing will not be covered by health insurance.
Previously, several attempts have been made to test for variants in archival formalin-fixed, paraffin-embedded (FFPE) tissue,8, 9 but until now, only the Ashkenazi Jewish founder mutations have successfully been tested for in FFPE samples.10, 11 However, outside the Ashkenazi Jewish community, testing for specific (founder) variants is insufficient for accurate risk assessment and counseling of families with increased risk of breast and ovarian cancer, and unknown BRCA status.
A new routine, high-throughput analysis to test archival FFPE samples of non-tumor tissue for germline variants in BRCA1 and BRCA2 was introduced using HaloPlex target enrichment (Agilent, Midlothian, Scotland/UK) and next-generation sequencing technology (Illumina, San Diego, CA, USA), to determine whether a deceased relative harbored a germline BRCA1 or BRCA2 variant known to affect protein function. The results of the initial validation study, including 32 samples, and the first clinical experience, including 201 samples from deceased relatives, with this new FFPE testing analysis are described here in detail.
Materials and methods
FFPE tissue samples
In the validation study, 32 FFPE samples of non-malignant tissue, from women with a known BRCA1 or BRCA2 variant or wild type (women previously tested for BRCA variants) were chosen from families known at the Department of Clinical Genetics, Vejle hospital. The samples were chosen to include a wide range of variant types (frameshift, missense, small indels, splice site and large deletions) and the age of the tissue ranged from 1 to 14 years. Tissue samples were included based on availability sufficient amount of non-malignant tissue (no small biopsies) and tissue containing a substantial amount of nuclei (eg, not fat tissue). The FFPE samples were investigated by an experienced pathologist and 9 × 15 μm FFPE tissue sections were cut. If a sufficient amount of DNA was not gained from 9 × 15 μm sections, further 9 × 15 μm sections were cut and DNA was extracted. A maximum of 18 × 15 μm sections were used in three samples (Val6, Val19 and Val22).
Furthermore, all information regarding tissue type, age of tissue and variant status was blinded to the technical and bioinformatic staff, and blinding was only lifted after disclosure of the final report of BRCA1/2 variants in the validation study.
In the clinical study, we used the best available tissue, evaluated by an experienced pathologist. If optimal tissue was not available, less optimal tissue was used (in four cases, tissue containing malignant cells were used). In some samples more than 9 × 15 μm sections were used to obtain sufficient amount of DNA for the analysis.
DNA extraction
DNA was extracted from three tubes each containing 3 × 15 μm FFPE tissue sections per sample using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to QIAamp DNA FFPE Tissue Handbook with a few modifications (www.qiagen.com). The DNA was validated using 1% Tris-acetate EDTA gel electrophoresis and DNA concentrations were determined using PicoGreen (Invitrogen, Life Technologies Europe BV, Nærum, Denmark). To verify overall quality of the DNA extracted from FFPE samples, the level of fragmentation was estimated using a quality control assay (QC assay) based on PCR provided by Agilent (Agilent Technologies, 2012). HapMap DNA (NA12878) sample was used as a non-degraded control (Coriell Institute). According to the results of the QC assay, samples were classified as good (26), medium (2) or poor (2), see Table 1. Furthermore, all DNA samples were analyzed on an Agilent TapeStation (Agilent Technologies) using Genomic ScreenTape and reagents according to Agilent gDNA ScreenTape System Quick Guide. To compensate for a higher level of fragmentation, the amount of input DNA for further analysis was 225, 500 and 1000 ng for the samples classified as good, medium or poor, respectively. A flowchart of the DNA quality assessment before NGS library preparation is shown in Figure 1.

Flowchart of FFPE DNA sample and QC assays. After DNA extraction, three QC assays were performed to validate the quality of the DNA: (1) QC-PCR was used to estimate the level of fragmentation by comparing two PCR products amplified from FFPE DNA with the amplified PCR products from HapMap DNA (NA12878). According to the results of the QC-PCR, samples were classified as good, medium or poor. (2) DNA concentrations were measured using a PicoGreen assay. (3) All DNA samples were analyzed on a TapeStation to view the fragmentation profile of the DNA. Either the profiled was rated as ‘flat’ indicating that DNA was highly degraded or not present, or the profile was rated as ‘peak’ indicating that the DNA was degrade but had a peak when looking at the electropherogram. If a sample was rated poor, had a DNA concentration less than 1 ng/μl and a ‘flat’ fragmentation profile, the DNA sample had failed QC. Only selected DNA samples failing QC were passed on to library preparation and sequencing, if there was a known variant in the family to search for.
HaloPlex target enrichment and sequencing
Twenty-nine genes encoding BRCA1/2 and other important proteins involved in the homologous recombination pathway,12, 13 were included in the HaloPlex (Illumina 100) custom design of 552 targets with a 118889-bp region of interest (ROI) (Agilent Technologies, 2012).14 The fraction of bases in ROI that can be analyzed covered 98.8% of the target region. HaloPlex libraries were constructed according to manufacturer’s protocol v.D4 (Agilent Technologies, 2013).15 Indexes were incorporated for each sample during enrichment, allowing samples to be multiplexed before sequencing. A total of 30 HaloPlex libraries were validated on a bioanalyzer High Sensitivity chip (Agilent Technologies).
After enrichment, HaloPlex libraries were diluted to 10 nM, pooled, denatured and subjected to paired-end (2 × 150 bp), single index (8 bp) reversible terminator based DNA sequencing on a MiSeq (Illumina).
In both parts of the study only data regarding BRCA1 and BRCA2 were analyzed.
Alignment
For each sequenced sample, the raw fastq files generated from the Illumina MiSeq system were trimmed with TrimGalore (version 0.33), subsequently mapped to the hg19 human reference genome using MOSAIK (version 2.2),16 and converted to BAM using Samtools (version 0.1.19).17 Each sample BAM file was preprocessed with Genome Analysis Toolkit 18, 19 (GATK version 3.1.1; local realignment around indels and base quality score recalibration), before variant calling. General alignment statistics (eg, number of aligned reads, size of insert fragment, etc) were generated with BAMtools (version 2.3.0).20 Target-specific alignment statistics (ie, per base-/region-/gene-/sample-coverage and coverage percentage of ROIs), were obtained using GATK DepthOfCoverage.
Variant calling and annotation
Following preprocessing of BAM files, variant calling was performed using GATK HaplotypeCaller (GATK version 3.1.1). Low-quality/false positive variants were filtered out using GATK VariantFiltration. Only variants fulfilling the following criteria FS<250 & QD>2.0 & QUAL>200 & HomopolymerRun (HRun)<7 & DP>10, were kept in the filtered single-sample variant call sets, which were subsequently merged to produce a multi-sample call set for all validation samples or clinical samples, respectively. Each merged call set was annotated using SnpEff (version 3.6)21 and VariantTools (version 2.3),22 using build-in and custom annotation tracks. NCBI reference sequences (RefSeq) NM_007294.3 and NM_000059.3 have been used for the annotation of BRCA1 and BRCA2 variants, respectively. These RefSeq transcripts are included in the Locus Reference Genomic (LRG) data LRG_292-BRCA1 and LRG_293-BRCA2. BRCA1/2 variant data has been submitted to Leiden Open Source Database at http://databases.lovd.nl/shared/individuals/PatientID (PatientID: 00051505–00051521). Analysis of coverage data and quality metrics were performed in R (version 3.0.2 Frisbee sailing),23 using base packages and the CRAN package ‘pheatmap’ for heatmap representation of coverage data.24 Heatmap clustering is based on complete linkage on Euclidian distances. Spearman’s Rho was used to assess the correlation between FFPE age and ROI coverage.
Deletion/duplication testing using MLPA
Clinical FFPE DNA samples were subjected to mulitiplex ligation-dependent probe amplification (MLPA). A total 5 μl of FFPE DNA was used for each MLPA reaction, and analysis was conducted according to manufactures one-tube protocol (MRC-Holland, Amsterdam, The Netherlands). SALSA MLPA P002 BRCA1 probemix and SALSA MLPA P045 BRCA2/CHEK2 probemix were used for the MLPA analysis of BRCA1 and BRCA2, respectively. Fragment separation was conducted on an ABI3130 using POP6 polymer and 36 cm capillaries. Injection mixture contained 0.5 μl MLPA PCR reaction and 12 μl Hi-Di formamide master mix (0.5 μl GS-500 MW marker+12 μl Hi-Di formamide). Run module: FragmentAnalysis; injection voltage: 1.4 kV; injection time 15 s; run voltage: 15 kV; run time: 2400 s; and oven temperature: 55 °C. The fragment analysis results were analyzed using GeneMapper and GeneMarker. The RefSeq transcripts NM_007294.3 and NM_000059.3 are included in the LRG data LRG_292-BRCA1 and LRG_293-BRCA2, respectively. The LRG-specific exon numbering for BRCA1 and BRCA2 has been used.
Ethical considerations
Since the validation study only involved a new method for finding known variants and no new knowledge about the participant’s genetic status was gained, we were allowed to perform the genetic investigations without prior consent from the subjects who participated with their tissue samples. This permission was granted by the Regional Committee on Health Research Ethics, Region of Southern Denmark. In the clinical setting, all investigations on tissue samples from deceased persons, was performed only after informed consent from a closely related family member seeking genetic counseling, according to standard practice in clinical genetics.
Results
Correct call of pathogenic variants in BRCA1 and BRCA2
DNA extraction was successful in 30 out of 32 FFPE samples from women with a known BRCA1/2 variant or wild-type. In two samples, the amount of DNA was too low (sample 11 and 17) to perform target enrichment, library preparation and sequencing. In 25 out of 30 sequenced samples of non-cancer FFPE tissue, it was possible to correctly identify and classify either a BRCA1 or BRCA2 variant (true positive: 20 samples) or wild type (true negative: five samples), resulting in an accuracy of 83.3%, see Table 1. In three samples (Val1, Val14 and Val26), it was not possible to identify a large intragenic deletion c.(80+1_81−1)_(4986+1_4987−1)del corresponding to the deletion of exons 3–15 in BRCA1. Furthermore, two samples did not result in a correct variant call, due to poor DNA quality, and hence a low coverage (9x) at the position of interest (Val29) and a skewed read distribution with 81% and 19% of read data supporting the reference and variant allele, respectively (Val15). Consequently, a false negative result was observed in a total of 5 of the 30 samples. However, the variant of Val15 was correctly called in the raw data, but was filtered out as a result of the skewed read distribution. No additional (ie, false positive) variants affecting protein function were identified (false discovery rate: 0.0). Albeit pertaining to a limited number of samples, these findings indicate that the method and analysis strategy used provides high sensitivity (0.8) and very-high specificity (1.0) and positive prediction value (1.0). In contrast, the five false negative calls and the inclusion of only five true negative samples results in a lower negative prediction value (0.5).
MLPA was not applied in the validation study.
Target performance: validation study
In this study, the ROI is defined as coding exons plus 20-bp flanking region. The ROI coverage was not uniform across the samples as 30 × coverage varied between 48.7 and 99.3% for all 29 target genes (All), 47.7–100% for BRCA1 and 41.4–99.7% for BRCA2, see Table 1. This is also illustrated in the heatmap, see Figure 2a, where an overall good coverage was found in 24 out of the 30 samples. In six samples, the coverage was more diverse across the 29 target genes; Val29 had the lowest 30x coverage percentage across all target genes, indicated by the blue color in the heatmap. Besides Val29, five samples (Val6, Val9, Val15, Val16 and Val19) had a low-medium (<65%) 30x coverage percentage across seven out of the 29 target genes. Common for these five samples was that the FFPE sample age was more than 10 years upon DNA purification. The age of FFPE samples varied from 1 to 14 years. A significant inverse correlation between the age of FFPE sample (years) and the percentage of ROIs with at least 30x coverage (ρ=−0.598, P<0.01) was detected, see Figure 2b. Nevertheless, some samples aged 10 or more years (Val12, Val18, Val24, Val26 and Val28) still resulted in a high percentage of ROIs (>80%) with at least 30x coverage. Furthermore, a strong correlation between the median fragment length (bp) sequenced and the percentage of ROIs with at least 30x coverage (ρ2=0.914, P<0.001) was detected as well.

(a) Heatmap of all 30 sequenced samples from the validation study. Red color represents that 100% of ROI is covered at least 30x times whereas blue color represents 0% coverage. Each column represents one sample and each row the gene sequenced. (b) Validation study: an inverse correlation between age of the sequenced archival FFPE sample and the percentage of 30x coverage of ROI, (ρ2=−0.598, P<0.01). Red dots represent correct call of variant status, gray dots represent incorrect calls.
Target performance: Clinical Samples
In the clinical data, the ROI coverage was not uniform across the samples as 30x coverage varied between 21.1 and 99.5% for all 29 target genes, 18.1 and 99.8% for BRCA1 and 15.1 and 99% for BRCA2. As seen in the validation study, a significant inverse correlation between age of FFPE sample (years) and the percentage of ROIs with at least 30x coverage ρ=−0.386 P<0.01) was detected. Even though some samples performed inadequate regarding ROI coverage, positive results were obtained in some of these samples, see Figure 3. As an example, a variant known to affect protein function in BRCA1 was found in sample D13-2662, even though the percentage of ROI with at least 30x coverage (all) was low (24.8%), see Table 2.

Clinical FFPE Samples: correlation between age of the 165 sequenced archival FFPE samples and 30x coverage of ROI, (ρ=−0.0386, P<0.01). Red dots represent a positive finding of a variant known to affect function or VUS in BRCA1/2, gray dots represent negative findings (no variants).
Clinical experience with 201 samples from deceased persons
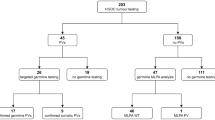
DNA was successfully extracted from 201 clinical FFPE samples from deceased relatives from families with a high suspicion of carrying a BRCA1 or BRCA2 variant, based on clinical experience or using the BOADICEA risk estimation program.25 The age of the samples ranged from 0 to 43 years. Based on the results of the QC assay, 23 samples were rejected for further analyses. The remaining 178 FFPE DNA samples were subjected to target enrichment library preparation. 13 samples were removed due to failed library preparation, and the remaining 165 FFPE samples, aged 0–38 years, were successfully sequenced and subjected to BRCA data analysis, see Figure 4. After BRCA data analysis, 15 out of the 165 samples were analyzed primarily to search for a known familial BRCA1/2 variant, by visual inspection of the known genomic position.

BRCA data analysis of 165 clinical FFPE samples: a total of 18 variants were detected in 17 out of the 150 FFPE samples with unknown BRCA-status. A total of three variants known to affect function and one variant likely to affect function in BRCA1, six variants known to affect function and one variant likely to affect function in BRCA2, four VUS in BRCA1 and three VUS in BRCA2 were detected. In the 15 samples analyzed because of a familial variant known to affect function in BRCA1/2 (or VUS), 13 variants were detected in 11 samples. A total of seven variants known to affect function in BRCA1 and two variants known to affect function in BRCA2, as well as one VUS in BRCA1 and three VUS in BRCA2 were detected. Number of samples are written in red, and number of variants are written in blue.
In the 150 FFPE samples, a total of three variants known to affect function, and one variant likely to affect function in BRCA1, six variants known to affect function, and one variant likely to affect function in BRCA2, four VUS in BRCA1 and three VUS in BRCA2 were detected (Table 2 shows all the described variants in detail). In the remaining 133 samples, no variants or only benign/likely benign variants were found. In the 15 samples analyzed because of a known familial variant (or VUS), seven variants known to affect function in BRCA1 and two variants known to affect function in BRCA2, as well as one VUS in BRCA1 and three VUS in BRCA2 were found (Table 3 shows all the described variants in detail).
In three samples more than one variant was found; in sample D14-1242, two VUS in BRCA1 c.1486C>T and c.5297T>A (HGVS) were identified, see Table 2. In sample D14-1243, a biopsy from the gastric mucosa harboring an adenocarcinoma, two variants in BRCA1 were found, see Table 3. The first, a familial variant known to affect BRCA1 function; c.427G>T (HGVS) was correctly identified and a second variant c.4043_4043delG (HGVS) was also detected, and is assumed to be of somatic origin. The second variant is not known in the Breast Cancer Information Core database, but it induces a frameshift leading to a premature stop codon. In sample D14-1837, a BRCA1 variant known to affect function c.427G>T was found together with a VUS in BRCA2 c.6287C>T, see Table 3.
MLPA analysis was used to detect larger intragenic deletions or duplications. The BRCA1 and BRCA2 MLPA results were normal in 90 and 80 samples, respectively, but non-informative because of low DNA quality in 87 and 97 samples, respectively. BRCA1 and BRCA2 MLPA analysis were not performed in one sample. No large deletions or duplications in BRCA1/2 were found in the clinical samples.
Discussion
To our knowledge, this is the first published successful attempt to systematically test archival FFPE samples of non-cancer tissue for germline variants in BRCA1/2. Previous attempts, using more ‘classical’ methods, such as single-strand conformation polymorphism analysis or sanger sequencing, resulted in a substantial rate of both false positive and false negative results8, 9 or at best was limited to search for the known Jewish Ashkenazi founder mutations.10, 11, 26 However, a reliable NGS method for detection of variants in BRCA1/2 in FFPE samples from tumor tissue was recently published.27
In the validation study, the variant calling resulted in a true match in 25 out of 30 sequenced samples (83%). It was not possible to detect a large intragenic deletion in BRCA1 in three samples (Val1, Val14 and Val26). However, this was expected because of the choice of sample preparation method, which is based on an amplicon target-enrichment technique with non-random DNA shearing, making copy number variation detection difficult, since duplicate reads cannot be identified. Based on this, it is recommended to use MLPA or a similar method for the detection of larger deletions or duplications, although analyzing highly degraded DNA may cause inconclusive results. Furthermore, it was not possible to correctly call a single base substitution (Val15) or a single base insertion (Val29) in two samples. However, after unblinding of the study, the variant of Val15 could be correctly called in the raw data, but was filtered out as a result of the skewed read distribution between wild type and alternative. By changing the settings in the data analysis pipeline, it was possible to correctly call this variant. In the case of Val29, the coverage was low (9x coverage), and the variant was not detected in any reads covering this position. Another important result is that we did not find any false positive variants, in either BRCA1/2-positive or in the wild-type group. Formalin fixation is known to introduce alterations in the DNA and this may result in false positive findings, which has been reported from earlier attempts.9
The DNA quality is a strong predictor of the outcome of the analysis, and a significant correlation between the median fragment length (bp) of the sequenced DNA and 30x coverage percentage across the ROI supports this observation. Highly degraded DNA contains shorter DNA fragments and therefore more DNA is required to obtain successful target enrichment. However, based on this study we recommend using 9 × 15 μm sections for DNA extraction. As seen from Figure 2, coverage is decreasing with age of the tissue (especially after 10 years of age), implying that DNA quality decreases with age, but with large variation, as some samples aged 10 or more years may still result in a high 30x coverage.
When implementing the test in our clinical setting, a greater variation in both age of the tissue, coverage and hence outcome of the sequencing was detected compared to the validation study. The age of the clinical samples was up to 43 years, but DNA extracted from tissue older than 38 years did not meet our quality criteria for library preparation and sequencing. The percentage of 30x coverage of the target genes varied more compared to the validation data set. Even though, the coverage is declining with increasing age, variants were detected in samples with coverage in the lower range, see Figure 3. These results represent the cohort tested with the current analytical set-up, and since no true information exists regarding incidence of BRCA1/2 mutations in this cohort, it is impossible to calculate neither sensitivity nor specificity of the test. The overall negative result from the MLPA analysis comes as no surprise, as deletions in BRCA1/2 are found in a minority (3.8%) of the BRCA1/2-positive families in Denmark.28
Detecting a variant known to affect function in BRCA1/2 in a FFPE sample should always prompt further investigation. A variant should always be verified in a new sample, either from a different tissue from the same person, or should be verified in another family member. Before a variant is verified, it can only be assumed positive, since there is a risk of false positive findings due to false positive calls from software, PCR amplification, sequencing error, or due to alterations in the DNA caused by the fixation or age.29 Negative results (normal sequence) should always be interpreted with caution. If the 30x coverage is high (we recommend >90%), the result may be interpreted, as if it was a blood sample from the same person. However, the possibility of a variant in the ROI that is not covered can never be excluded. If coverage is low, risk estimation should be offered, as if no analysis was performed and based solely on the family history. In families where a VUS is found, FFPE testing could potentially be used to perform segregation analysis to analyze, whether the variant co-segregate with the disease in a family. FFPE testing could also be used to investigate if a variant was inherited from the maternal or paternal side of the pedigree. This could potentially reduce anxiety and the economic burden of testing family members on both sides of a family, instead of testing only the relevant side, after identifying the variant in either the mother or father (if a tissue sample is available).
Testing FFPE samples may be used for other purposes than BRCA1/2 testing. As the percentage of 30x coverage across the 29 target genes in the panel used, is generally consistent, the method can be used to test for variants in other highly penetrant breast and ovarian cancer genes (like PALB2, RAD51C/D, PTEN, CDH1 and TP53). The usefulness of the test is anticipated to increase with mortality risk of the investigated gene. If there is a high mortality rate in carriers of, eg, TP53 or CDH1 variants, it will be less likely that there is a living carrier to investigate. We used normal tissue, in order to search for germline variants, but the method could also be applied to malignant tissue, in order to search for somatic variants in genes involved in the homologous recombination pathway (the BRCAness genes).30 Finding somatic variants in one of these genes could be important for future management of cancer with a potential of being targets for treatment with PARP inhibitors. The recently described Poly ADP-Ribose Polymerase (PARP)-inhibitors have in several studies showed promising results in treating cancer in carriers of BRCA1/2 variants known to affect function.31, 32, 33
Future development of the FFPE testing includes improving the design of the HaloPlex probes. Increasing the capture and amplification of smaller DNA fragments will improve the coverage especially in degraded DNA samples. Optimizing the DNA extraction could also improve the outcome, as higher DNA yield and concentration may lead to more usable DNA.
In conclusion, testing deceased persons for variants in BRCA1/2, using HaloPlex target enrichment and next-generation sequencing, is possible in archived FFPE tissue samples aged up to 30 years and may help to more accurately evaluate the risk of breast and ovarian cancer in some families, where genetic counseling otherwise would rely on risk assessment based on family history alone.
References
Miki Y, Swensen J, Shattuck-Eidens D et al: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266: 66–71.
Wooster R, Bignell G, Lancaster J et al: Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789–792.
Antoniou A, Pharoah PD, Narod S et al: Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72: 1117–1130.
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE : Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343: 692–695.
Breast Cancer Linkage C: Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 1999; 91: 1310–1316.
Metcalfe KA, Birenbaum-Carmeli D, Lubinski J et al: International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer 2008; 122: 2017–2022.
Cruger DG, Kruse TA, Gerdes AM : 'Indirect' BRCA1/2 testing: a useful approach in hereditary breast and ovarian cancer families without a living affected relative. Clin Genet 2005; 68: 228–233.
Bernstein JL, Thompson WD, Casey G et al: Comparison of techniques for the successful detection of BRCA1 mutations in fixed paraffin-embedded tissue. Cancer Epidemiol Biomarkers Prev 2002; 11: 809–814.
Wong C, DiCioccio RA, Allen HJ, Werness BA, Piver MS : Mutations in BRCA1 from fixed, paraffin-embedded tissue can be artifacts of preservation. Cancer Genet Cytogenet 1998; 107: 21–27.
Zhang L, Kirchhoff T, Yee CJ, Offit K : A rapid and reliable test for BRCA1 and BRCA2 founder mutation analysis in paraffin tissue using pyrosequencing. J Mol Diagn 2009; 11: 176–181.
Adank MA, Brogi E, Bogomolniy F et al: Accuracy of BRCA1 and BRCA2 founder mutation analysis in formalin-fixed and paraffin-embedded (FFPE) tissue. Fam Cancer 2006; 5: 337–342.
Turner N, Tutt A, Ashworth A : Hallmarks of 'BRCAness' in sporadic cancers. Nat Rev Cancer 2004; 4: 814–819.
Rigakos G, Razis E : BRCAness: finding the Achilles heel in ovarian cancer. Oncologist 2012; 17: 956–962.
HaloPlex Target Enrichment from FFPE Tissues, Application note: Agilent Technologies, 2012 Available from www.agilent.com/genomics/ngs.
HaloPlex Target Enrichment System – For Illumina Sequencing – Protocol version D.4: Agilent Technologies 2013, Available from www.agilent.com/genomics/ngs.
Lee WP, Stromberg MP, Ward A, Stewart C, Garrison EP, Marth GT : MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One 2014; 9: e90581.
Li H, Handsaker B, Wysoker A et al: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
McKenna A, Hanna M, Banks E et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
DePristo MA, Banks E, Poplin R et al: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT : BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011; 27: 1691–1692.
Cingolani P, Platts A, Wang le L et al: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012; 6: 80–92.
San Lucas FA, Wang G, Scheet P, Peng B : Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics 2012; 28: 421–422.
Team RC : A Language and Environment for Stastistical Computing. R Foundation for Stastistical Computing: Vienna, 2013.
Kolde R, Pheatmap Pretty Heatmaps. R package version 0.7.7 2013.
Lee AJ, Cunningham AP, Kuchenbaecker KB et al: BOADICEA breast cancer risk prediction model: updates to cancer incidences, tumour pathology and web interface. Br J Cancer 2014; 110: 535–545.
Mangold KA, Wang V, Weissman SM, Rubinstein WS, Kaul KL : Detection of BRCA1 and BRCA2 Ashkenazi Jewish founder mutations in formalin-fixed paraffin-embedded tissues using conventional PCR and heteroduplex/amplicon size differences. J Mol Diagn 2010; 12: 20–26.
Ellison G, Huang S, Carr H et al: A reliable method for the detection of BRCA1 and BRCA2 mutations in fixed tumour tissue utilising multiplex PCR-based targeted next generation sequencing. BMC Clin Pathol 2015; 15: 5.
Thomassen M, Gerdes AM, Cruger D, Jensen PK, Kruse TA : Low frequency of large genomic rearrangements of BRCA1 and BRCA2 in western Denmark. Cancer Genet Cytogenet 2006; 168: 168–171.
Quach N, Goodman MF, Shibata D : In vitro mutation artifacts after formalin fixation and error prone translesion synthesis during PCR. BMC Clin Pathol 2004; 4: 1.
McCabe N, Turner NC, Lord CJ et al: Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006; 66: 8109–8115.
Fong PC, Boss DS, Yap TA et al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123–134.
Gelmon KA, Tischkowitz M, Mackay H et al: Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 2011; 12: 852–861.
Ledermann J, Harter P, Gourley C et al: Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 2014; 15: 852–861.
Acknowledgements
We thank Tinna Herløv, Department of Clinical Pathology, Vejle Hospital and Kaja Skovgaard Jensen, Department of Clinical Genetics, Vejle Hospital for their expert technical assistance cutting the FFPE tissue sections and in the laboratory with the DNA extraction, HaloPlex library preparation and sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Petersen, A., Aagaard, M., Nielsen, H. et al. Post-mortem testing; germline BRCA1/2 variant detection using archival FFPE non-tumor tissue. A new paradigm in genetic counseling. Eur J Hum Genet 24, 1104–1111 (2016). https://doi.org/10.1038/ejhg.2015.268
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.268
This article is cited by
-
Ovarialkarzinom: Molekularpathologie und molekular gesteuerte Therapie
Die Pathologie (2023)
-
Germline FFPE inherited cancer panel testing in deceased family members: implications for clinical management of unaffected relatives
European Journal of Human Genetics (2021)
-
Targeted capture-based NGS is superior to multiplex PCR-based NGS for hereditary BRCA1 and BRCA2 gene analysis in FFPE tumor samples
BMC Cancer (2019)
-
Identification of novel mutations in FFPE lung adenocarcinomas using DEPArray sorting technology and next-generation sequencing
Journal of Applied Genetics (2018)
