
Abstract
The global demand for products that effectively prevent the development of male-pattern baldness (MPB) has drastically increased. However, there is currently no established genetic model for the estimation of MPB risk. We conducted a prediction analysis using single-nucleotide polymorphisms (SNPs) identified from previous GWASs of MPB in a total of 2725 German and Dutch males. A logistic regression model considering the genotypes of 25 SNPs from 12 genomic loci demonstrates that early-onset MPB risk is predictable at an accuracy level of 0.74 when 14 SNPs were included in the model, and measured using the area under the receiver-operating characteristic curves (AUC). Considering age as an additional predictor, the model can predict normal MPB status in middle-aged and elderly individuals at a slightly lower accuracy (AUC 0.69–0.71) when 6–11 SNPs were used. A variance partitioning analysis suggests that 55.8% of early-onset MPB genetic liability can be explained by common autosomal SNPs and 23.3% by X-chromosome SNPs. For normal MPB status in elderly individuals, the proportion of explainable variance is lower (42.4% for autosomal and 9.8% for X-chromosome SNPs). The gap between GWAS findings and the variance partitioning results could be explained by a large body of common DNA variants with small effects that will likely be identified in GWAS of increased sample sizes. Although the accuracy obtained here has not reached a clinically desired level, our model was highly informative for up to 19% of Europeans, thus may assist decision making on early MPB intervention actions and in forensic investigations.
Similar content being viewed by others


Introduction
Male-pattern baldness (MPB) or androgenic alopecia is the most common type of hair loss in men, with a prevalence of around 20% at age 20–30, and the incidence growing at 10% per decade.1, 2 MPB is a chronic problem commonly seen by dermatologists,3 with severe psychosocial consequences4 and limited effective therapeutic options.5 The effectiveness of most treatments (eg, minoxidil or finasteride) relies on how early they are applied. Therefore, the ability to predict the early-onset or normal MPB status using DNA variants may have important implications for treatment strategies. Furthermore, owing to its widespread prevalence and the fact that most criminals are men, MPB in principle could help identify unknown perpetrators via the concept of forensic DNA phenotyping,6, 7 especially in light of the current progress in predicting chronological age from DNA data.8, 9 However, so far there is no established genetic model for predicting MPB from genetic data providing motivation for the present study.
MPB is a highly heritable visible trait, with estimated heritability of about 80% in young2 and elderly males.10 A locus on chromosome Xq12 harboring the androgen receptor gene (AR) and its neighboring ectodysplasin A2 receptor gene (EDA2R) is known as the major locus for MPB.11, 12 In addition, two genetic loci on chromosome 20p11 (PAX1/FOXA2) and 7p21.1 (HDAC9) were identified to be involved in MPB.13, 14, 15 A meta-analysis of seven GWASs for early-onset MPB involving ~13 000 individuals of European origin conducted by the International MAAN Consortium16 replicated these loci and highlighted five additional loci showing genome-wide significant association with MPB; these included 1p36.22, 2q37.3, 7q11.22, 17q21.31, and 18q12.3. Individuals in the highest-risk quartile of a genotype score had an approximately sixfold increased risk of early-onset MPB. In addition, the most recent study by Heilmann et al17 comprising 2759 cases and 2661 controls successfully identified four additional autosomal loci showing genome-wide significant association with MPB; these included 2q35, 3q25.1, 5q33.3, and 12p12.1 (identifying a total of 12 genetic loci so far). These findings on one hand highlight a relatively strong X-linked major gene locus effect (odds ratio per risk allele ~2.5 at the AR locus) and on the other hand suggest a highly polygenetic autosomal component (odds ratios at individual loci <1.5). Here we estimate to what degree MPB is predictable using the currently known DNA markers discovered from GWAS so far. This study includes a total of 2725 German and Dutch males, with included early-onset MPB patients and controls as well as middle-aged and elderly cases and controls.
Materials and Methods
Ethics statement
All studies were approved by the institutional ethics review committees at the relevant organizations, and written informed consent was provided by all participating individuals.
Rotterdam Study
The Rotterdam Study (RS) is a population-based prospective study of Dutch elderly subjects (>45 years of age) consisting of an initial cohort and two extensions.18 MPB status was assessed by trained physicians according to the Norwood–Hamilton grading scale1, 19 with grades 1–12. Cases were defined as grade IV–VIII and otherwise controls. The current study included 1161 male RS subjects. RS samples have not been used for MPB GWAS before and are not part of MAAN; hence, RS sample are completely independent from previous MPB SNP discoveries. Anonymized individual-level phenotype and genotype data used for the prediction analysis from the Rotterdam Study participants are available in Supplementary Table S1.
Erasmus Rucphen Family Study
Erasmus Rucphen Family (ERF) is a family-based study that includes inhabitants of a genetically isolated community in the south–west of the Netherlands, studied as part of the Genetic Research in Isolated Population program.20 Study population includes ~3000 individuals who are living descendants of 22 couples who had at least six children baptized in the community church. All data were collected between 2002 and 2005. The population shows minimal immigration and high inbreeding. Cases were defined as Norwood–Hamilton grading scale1, 19 IV–VIII at any age or grade II–III between 50 and 60 years of age or grade I before 50 years of age, and controls otherwise. The current study included 567 male ERF subjects. ERF samples have not been used for MPB GWAS before and are not part of MAAN; hence, ERF samples are completely independent from previous MPB SNP discoveries. The ERF Study data are archived in European Genome–Phenome Database (EGA) with the accession code EGAS00001001134.
BONN Study
The hair status of each participant was assessed by a dermatologist according to the Hamilton/Norwood (HN) classification.1, 3 Affected men were aged <30 years with HN grades IV–VII, or <40 years with HN grades V–VII, and were thus representative of the most severely affected 10% for the respective age classes (N=581). The control sample comprises 270 men aged ⩾60 years with no signs of AGA (20% least affected individuals in the population) and 146 male controls (HN⩽V) that were recruited as part of the Heinz Nixdorf Recall cohort (risk factors, evaluation of coronary calcium and lifestyle) at the University of Essen. All the cases and controls were of German descent.13 Note that the 581 cases and the 146 controls from the BONN Study used here were part of the initial MAAN study,16 and all BONN subjects were also part of the previous study of Heilmann et al,17 based on which the SNPs used here for MPB prediction were initially discovered. There is thus a potential risk in overestimating the prediction accuracy in BONN but at most by only a small degree as BONN was only a small component of the MAAN study16 (ie, 14.9% of cases and 1.6% of controls in MAAN and 21.1% of cases and 15.6% of controls in the Heilmann study). The RS and ERF subjects are completely independent from previous studies. The BONN Study data are archived in European Genome–Phenome Database (EGA) with the accession code EGAS00001001354.
Genotyping and quality control
The genotyping platforms, quality controls, and imputation methods used in participant studies have been described in detail previously. In brief, extensive quality control thresholds were applied to include common SNPs (minor allele frequency >1%) with a high call rate (95%) for genotyped SNPs; SNPs demonstrating deviation from Hardy–Weinberg equilibrium (P<10−6) were excluded. SNP genotypes from all cohorts were imputed using the 1000-Genome Project as the reference panel with high-quality metrics (variance ratio 0.3 for MACH and proper info statistic 0.4 for IMPUTE).21, 22 The X-chromosome in ERF was imputed using HapMap-CEU samples as the reference. After all quality controls, this study includes 2 444 603 autosomal SNPs and 239 421 X-chromosome SNPs in RS; 2 266 959 autosomal SNPs and 89 191 X-chromosome SNPs in BONN; and 6 099 730 autosomal SNPs and 16 708 X-chromosome SNPs in ERF. All SNPs are described using dbSNP ID according to human reference assembly GRCh37.p13.
Statistical analyses
We initially selected 20 SNPs from 12 genomic loci for prediction analysis from the Table 2 and the Supplementary Table S2 of Li et al16 as well as Table 1 of Heilmann et al.17 A candidate SNP analysis was conducted using logistic regression in all three cohorts for the 20 SNPs assuming additive allele effect adjusted for age at examination when appropriate. As the AR locus has a relatively large effect, we additionally selected four SNPs from the AR locus showing some residual effect based on a conditional logistic regression analysis of all SNPs within 66.5–67.9 Mbp of the AR locus in a stepwise manner, that is, until the newly included SNP is not significant anymore at P<0.05 level in a multivariate analysis of all SNPs accumulated in previous steps. A prediction analysis including all 24 selected SNPs was conducted separately in all three cohorts; age at examination was included as a predictor in RS and ERF but not in BONN consisting of early-onset cases and screened elderly controls. Then the final selection and ranking of the SNP predictors was based on stepwise analysis of the Akaike information criterion23 using R function ‘step’. The prediction models were trained separately in the three cohorts based on multivariate logistic regression. Because our sample size is not large and an explicit validation set was not available, we used the leave-one-out cross-validation method to prevent overfitting, that is, the prediction models were trained in all subjects except one, who was used for validation by applying the trained model on this subject. We repeated this procedure iteratively over the whole cohort by leaving each subject out, and obtained the predicted probability of baldness status for all subjects. The predicted probabilities are compared with observed baldness status, where the AUCs24 were derived as an overall measurement of prediction accuracy. An AUC value ranges from 0.5 representing random prediction to 1.0 representing perfect prediction. Binary prediction of baldness status for each subject was defined if the predicted probability is >0.5 otherwise non-bald. The predicted and observed baldness status was compared using a confusion table, where sensitivity and specificity values are derived, both ranging from 0 to 1. Sensitivity and specificity values are a pair of inseparable accuracy parameters for a binary classifier; a perfect classifier would be described as 100% sensitive and 100% specific. All candidate SNP analysis and prediction analysis were conducted in R version 3.2.0 (http://www.r-project.org/).
Estimates of the proportion of variance explained were calculated using the Genome-wide Complex Trait Analysis (GCTA) tool v1.24 (http://gump.qimr.edu.au/gcta/).25 Genetic relationships were estimated using all autosomal SNPs (—make-grm, —maf 0.03). The top 10 eigenvectors from PCA analysis (—pca) were then used as covariates in a restricted maximum likelihood analysis (—reml) to estimate the proportion of the variance explained by SNPs (VG/VP or narrow-sense heritability h2), repeated for all autosomes as a whole, each autosome separately, and the X chromosome.
A GWAS for MPB status was separately conducted in all cohorts using logistic regression considering age at examination as a covariate (except in BONN, which used early-onset cases) using PLINK1.9 beta.26 Family relationship was adjusted using first four principal components from EIGENSTRAT27 analysis. A meta-analysis of GWAS results was conducted using inverse variance fixed-effect analysis. P-values equal to or smaller than 5 × 10−8 were considered as genome-wide significant.
Results
The characteristics of study subjects (all male) are summarized in Supplementary Table S2 (BONN: 581 early-onset cases and 416 controls; RS: 619 cases and 542 controls; and ERF: 252 cases and 315 controls). The BONN data have been described in a previous GWAS,13 whereas RS and ERF data have not been used before for the genetic investigation of male-pattern baldness. Note that the BONN Study included only early-onset MPB cases (<40 years of age) and screened elderly controls while the RS (mainly elderly individuals: mean age 67.79 years, min 51.54, max 96.73) and ERF (mainly middle-aged: mean age 49.05 years, min 18.07, max 79.03) are population-based studies without sample selection based on MPB status. The difference in MPB prevalence observed between RS (53.3%) and ERF (44.4%) is fairly consistent with a ~10% increase per decade of MPB1 (Supplementary Table S2).
Candidate SNP analysis
We focused on 20 candidate SNPs from 11 autosomal loci and the AR/EDA2R locus on X chromosome previously reported by Li et al16 and Heilmann et al17 (Table 1). Part of the BONN subjects was used in Li et al and Heilmann et al, so as expected, the allelic effects in BONN for all tested SNPs was consistent with previous reports and most of the tested SNPs showed significant association with MPB (P<0.05). In RS and ERF, the risk alleles previously reported in other populations also showed higher frequencies in cases than in controls for all SNPs tested, that is, OR>1.0, and seven SNPs at five genetic loci were nominally significantly associated with MPB (chr1p36: rs12565727 P=2.71 × 10−4; chr7q11: rs6945541 P=0.039; chr17q21: rs17762954 P=0.014 and rs12373124 P=5.18 × 10−3; chr18q12: rs10502861 P=0.036; chrXq12: rs1511061 P=2.16 × 10−11; and rs2497938 P=1.38 × 10−12; Table 1). In ERF, the allelic effect for all tested SNPs was consistent with previous studies and five SNPs at four genetic loci showed significant association with MPB (chr1p36: rs12565727 P=1.37 × 10−3; chr7q11: rs2073963 P=9.63 × 10−4; chr7q11: rs6945541 P=0.037; chrXq12: rs1511061 P=4.98 × 10−5; and rs2497938 P=6.38 × 10−5). The genetic associations at 2q37 and 20p11 were not statistically significant associated in both RS and ERF, likely due to the smaller sample sizes compared with the previous GWASs. In the present meta-analysis of the three cohorts, the only locus that did not show significant association was 3q25.1, but the allelic effect was in the same direction as Li et al. Overall, the observed allele effect sizes were similar between RS and ERF, and similar to previous estimates in other populations.14, 15, 16 Noticeable exceptions were: (1) the chromosome 20p11 SNPs showed much smaller effects in RS and ERF (eg, rs6047844 OR=1.09 in RS and 1.18 in ERF, Table 1) than previous estimations (OR=1.60 in Li et al and 1.82 in BONN); and (2) the SNPs at the EDA2R/AR locus showed much larger effect (rs1511061 OR=9.07) in BONN than in RS and ERF (OR 2.68–2.75). These differences are likely explained by the extreme design in BONN, as BONN included only early-onset MPB cases (<40 years of age) and a set of super controls (⩾60 years, no signs of MPB) while no MPB pre-selection was made in RS and ERF, in which the majority of the subjects were elderly people.
Prediction analysis
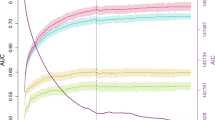
A prediction analysis included all 20 SNPs listed in Table 1 as predictors. We also added four extra X chromosome SNPs within a 66.5–67.9-Mbp region of the AR locus (rs113043121, rs471205, rs141476270, and rs182829063), which showed significant residual effect after conditioning on the top-associated X SNPs based on a stepwise conditional analysis (Table 2). For predicting MPB in RS and ERF we also added age as additional predictor (which in BONN was left out because of an extreme case–control design, that is, early-onset MBP patients and elderly screened controls). The overall prediction accuracy was derived using receiver-operating characteristic curves generated separately in the three studies (Figure 1a). In BONN, the AUC was estimated at 0.74 (sensitivity=0.84, specificity=0.50; Table 2) using 14 DNA markers and adding additional markers did not improve the prediction accuracy. In RS, the maximal AUC value was estimated at 0.71 (sensitivity=0.73, specificity=0.58) when age and 11 DNA markers were included (Table 2). In ERF, AUC was estimated at 0.69 (sensitivity=0.60, specificity=0.69) when age and 6 DNA markers were included (Table 2). Note that in ERF, some SNPs on chr2q35, chr17q21, and the X chromosome were not available, which could explain a lower accuracy in ERF than in RS. The extra SNPs were not available in ERF due to differences in the imputation procedures on the X chromosome (see Materials and Method section). The slightly lower prediction accuracies in RS/ERF versus BONN are likely explained by the fact that extreme early-onset MPB cases were selected in BONN, while RS and ERF are population-based cohorts without early-onset MPB selection. As may be expected, chronological age alone was the strongest predictor of MPB in both population-based sample sets (RS AUC=0.67, ERF AUC=0.62). Excluding the extra chrX-SNPs reduced the prediction accuracy in both RS and BONN, but only marginally (BONN AUC=0.73, RS AUC=0.71). Although the AUC values are lower than a clinically desired level (0.85) for diagnosis, our prediction model provided very accurate prediction results for a good proportion of individuals, that is, individuals with predicted probabilities of baldness <0.2 or >0.8 (BONN 8.2% <0.2 and 10.9% >0.8, Figure 1b; RS 5.2% <0.2 and 8.3% >0.8, Figure 1c; ERF 7.2% <0.2 and 0.9% >0.8, Figure 1d; also see Supplementary Table S3). In practice, our model may provide a highly informative test for these individuals (19% in BONN, 14% in RS, and 8% in ERF) but less informative for the rest.

Prediction results of male-pattern baldness in the three study populations. (a) Receiver-operating characteristic curves for predicting male-pattern baldness in three European population samples (BONN, RS, and ERF). In BONN Study the prediction model for early-onset MPB included 14 SNPs as predictors (Table 2); the model for predicting MPB in elderly RS people included 11 SNPs and age as predictors (Table 2); and the prediction model in ERF included 6 SNPs and age as predictors (Table 2). (b) Histogram of predicted probability overplayed with percentage of baldness in each probability bin (BONN Study). (c) Histogram of predicted probability overplayed with percentage of baldness in each probability bin (Rotterdam Study). (d) Histogram of predicted probability overplayed with percentage of baldness in each probability bin (ERF Study).
GCTA analysis
A GCTA analysis was performed to estimate the proportion of MPB variance explained by all common SNPs available in BONN and RS (Table 3). ERF was excluded from this analysis as it is a family-based study and it has a limited number of available X-chromosome SNPs. In BONN, the variance in liability to early-onset MPB was partitioned 55.8% to all autosomal common variants and 23.3% to X-chromosome variants. In RS, the variance was partitioned 42.4% to all autosomes and 9.8% to the X chromosome. Not surprisingly, early-onset MPB in BONN demonstrated an overall higher heritable component explainable by common DNA variants than normal MPB status in elderly individuals in RS. We further partitioned the proportion of variance explained by variants to each chromosome (Table 3,Supplementary Figure S1). In BONN, chromosomes 1 (h2=13.9%), 6 (12.5%), 17 (9.6%), 20 (24.3%), and X (27.2%) all explained a significant portion of early-onset MPB variance (Supplementary Figure S1A); the variance partitioned to other chromosomes was inconclusive due to large standard errors as the 95% confidence intervals included 0. In RS, only chromosome X showed a significant effect (h2=9.8%, 95% interval: 0.2–19.4%, Supplementary Figure S1B) in explaining MPB status in elderly individuals.
Meta-analysis of GWAS
GWASs for MPB were conducted in BONN, RS, and ERF. The QQ and Manhattan plot in ERF looks very different from RS and BONN (Supplementary Figure S2), that is, in ERF no genome-wide significant associations was detected including the EDA2R/AR locus (rs1511061 P=7.2 × 10−5). This underlies the importance of large sample sizes in GWAS even for detecting major gene effects. A meta-analysis of the GWAS results in the three cohorts identified 946 SNPs in four genetic loci that showed genome-wide significant association with MPB (P<5 × 10−8, Supplementary Table S4, Supplementary Figure S2). These include 739 SNPs on chromosome Xq12 between the EDA2R and AR genes, 205 SNPs on chromosome 20p11 (min P=1.76 × 10−15 observed for rs6075850), one SNP on chromosome 7p21.1 (rs756853 P=3.3 × 10−8), and one SNP on chromosome 6p25.1 (rs4959410 P=3.35 × 10−8). Except 6p25.1, all loci have been previously associated with MPB.14, 15, 16 The associated SNP at 6p25.1 is a single genome-wide significant finding not supported by additional SNPs in the region and thus warrants further validation.
Discussion
Candidate DNA variants in 12 previously discovered loci showed robust association with early-onset, as well as normal, MPB in German and Dutch males. Although one locus (3q25.1) was not statistically significantly associated, overall genetic effect sizes from all loci were similar to previous estimates. A major gene effect on Xq12 was confirmed with an allelic OR of up to 9.07 for early-onset MPB and 2.7–2.8 for normal MPB in middle-aged and elderly men. The combined predictive power of all candidate DNA variants was also higher for predicting early-onset MPB cases (AUC 0.74) than predicting MPB in middle-aged and elderly men from population-based studies (AUC 0.68–0.71), for whom age served as the strongest predictor (age-alone AUC 0.62–0.67). Although the overall predictive accuracy has not reached a practically desired level (>0.85), our risk model may prove useful in assisting decision making on early preventive actions for MPB and in forensic investigations for about 8–19% European individuals. Contrasting the observation that the 12 known DNA loci together explained rather limited variation of MPB liability, a variance partitioning analysis demonstrated that a large proportion of the phenotypic variance can be explained by all genotyped common SNPs available in the microarray data sets. This gap is likely due to many common variants with small effect sizes, which will likely be identified in future larger studies. Finally, our meta-analysis of the GWAS results identified one new locus at 6p25.1, which demonstrated significant association with MPB.
It has long been known that DNA variants in or near the AR gene increase the risk of patterned hair loss in both men and women, but the exact identity of the causal DNA variant(s) remains unclear. A common synonymous coding variant rs6152 G>A (StuI restriction site) in the exon 1 of the AR gene has been associated with MPB in previous studies.11, 12 For example, Ellis et al11 found that in an Australian cohort the G allele was present in 98.1% of young bald men, 92.3% of older bald men, and only 76.6% of non-bald men. The variant rs6152 is available in RS and was also highly significantly associated with MPB (P=5.6 × 10−8) but much less so than the top-associated X-chromosomal SNP rs1511061 (P=2.6 × 10−11 in RS), and it became nonsignificant (P>0.05) when conditioning on the genotypes of rs1511061. The SNP rs1511061 is an intergenic noncoding SNP located 236 kbp upstream of the AR coding region. These results suggest noncoding sequences upstream of AR containing functional variants. These could have regulatory effects as we recently established for noncoding variants in another human appearance trait – pigmentation.28, 29 The T allele (or A allele on the reverse strand of the genome) of rs1511061 is the major allele in our sample with a pronounced frequency in MPB cases (see Table 1). Assuming MPB is a monogenic phenotype caused by a single variant (ie, rs1511061), it would suggest baldness is the default phenotype in advanced ages, that is, the majority of males (wild-type allele) will eventually develop baldness while the minor allele provides a protective effect. The T allele of rs1511061 is also the major allele in HapMap-CEU (n=226, f=0.885), fixed in HapMap-HCB (n=90, f=1.0) and JPT (n=88, f=1.0), but reversely fixed in HapMap-YRI (n=120, f=0.0). This contradicts rs1511061 (and its high LD SNPs such as rs2497938) being causal because such a pattern of allele frequency distribution can hardly be correlated to the prevalence pattern at a global level as Asians and African–Americans have lower MPB prevalence and less severe MPB than Europeans.30, 31 Functional analyses of the noncoding sequence in this region, such as those we previously carried out for pigmentation gene regions,28, 29 are required to reveal the exact identity of the causal MPB genetic variant(s).
It has been suggested that there might be different genetic influences on balding in young men (ie, early-onset MPB) and on non-balding in elderly men, based on the observation of an increased frequency of non-balding in the fathers of non-bald elderly men.32 However, twin studies have shown that the heritability of MPB estimated in young men2 was similar to that estimated in elderly men10 (both around 80%). In our study, the genetic component of early-onset MPB (ie, BONN) showed somewhat different signature than that of normal MPB in elderly people (ie, RS), that is, the variance partitioning analysis suggests that a higher percentage of variance in early-onset MPB can be addressed by all common SNPs compared with that in elderly men. This is likely due to the extreme case–control design used in BONN Study, where early-onset cases and screened elderly controls were contrasted to enhance the genetic contrast. Furthermore, it is uncertain whether this discrepancy is due to phenotyping errors, as classifying MPB status in elderly people is much more error prone than defining early-onset cases. The variance partitioning analysis might be more sensitive to measurement errors than in twin studies, whereas for the latter even the effect of some differential misclassifications can be canceled out between monozygotic and dizygotic twins, that is, if the classification for one twin is differentially biased due to an unobserved factor, the same bias likely occurs to the other twin at a similar degree. Nevertheless, it is at least clear that the allelic effects at Xq12 and 20p11 are pronounced in early-onset MPB cases. For example, in our data the risk allele at Xq12 was presented in 96.5% of the early-onset cases, in 88% of the middle-aged and elderly cases, and in 75% of all controls.
The genetic architecture of human complex traits differs substantially even between the ones with similar heritability. For example, eye color and adult body height both have heritability estimates of about 80%. The genetic architecture of eye color, however, is relatively simple with a very strong major gene effect provided by a noncoding SNP at the HERC2 gene regulating transcription of the neighboring OCA2 pigmentation gene,29 and several minor-effect SNPs, which together predict the phenotype at very high accuracy (AUC>0.9 for blue/brown, explaining over 50% phenotypic variance).33, 34 On the other hand, adult body height has long served as a model trait of extreme genetic complexity, that is, without any major gene effect, 180 SNPs together could predict height at AUC of 0.75 and explain ~12% of the phenotypic variance,35 and with recent progress36 the AUC for predicting height is expected to be larger. In light of the current and previous studies, MPB appears something in the middle. It does have a major gene effect, that is, on the X chromosome (although much weaker than that of eye color) and likely involves many common variants with small effects (probably more than eye color). Improving the prediction accuracy for MPB will rely on the identification of more trait-associated DNA variants in GWAS of increased sizes, which will be a continuous and accumulative effort but certainly achievable in the future, as scientists already have achieved in studying human height (accumulative sample size over 250 000, several thousand SNPs together explain about 30% of the phenotypic variance).36 Therefore, we may expect that the prediction accuracy of MPB will eventually surpass that of body height given it has a known major gene effect, which is absent for height.
Our meta-analysis identified one new locus on chromosome 6p25.1 showing significant association with MPB. The associated SNP rs4959410 is surrounded by a pseudogene (BTF3P7, basic transcription factor 3 pseudogene 7) and several uncharacterized genes. The closest known genes are LY86 (lymphocyte antigen 86) and RREB1 (ras responsive element-binding protein 1). No existing evidence supports the involvement of any of these genes in hair loss. Therefore, this finding still needs to be confirmed in independent samples.
In conclusion, by taking all available SNPs previously found to be associated with MPB at a genome-wide significant level and testing their predictive value in 2725 German and Dutch males with early-onset MPB patients as well as from middle-aged to elderly men, we achieved prevalence-adjusted prediction accuracies expressed as AUC values of around 0.7 (where 0.5 means random prediction and 1.0 means completely accurate prediction). Although the prediction accuracy has not reached a level useful in practice, our preliminary genetic model may already assist decision making on early MPB preventive actions and in forensic investigations. Furthermore, our results imply that with more genome-wide significantly associated SNPs identified in the future and included in the prediction model together with the DNA markers presented here, male-pattern baldness will likely become predictable from DNA with high enough accuracy to allow routine practical applications such as in medicine and forensics.
Accession codes
References
Norwood OT : Male pattern baldness: classification and incidence. South Med J 1975; 68: 1359–1365.
Nyholt DR, Gillespie NA, Heath AC, Martin NG : Genetic basis of male pattern baldness. J Invest Dermatol 2003; 121: 1561–1564.
Hamilton JB : Patterned loss of hair in man; types and incidence. Ann N Y Acad Sci 1951; 53: 708–728.
Cash TF : The psychological effects of androgenetic alopecia in men. J Am Acad Dermatol 1992; 26: 926–931.
Sinclair R : Male pattern androgenetic alopecia. BMJ 1998; 317: 865–869.
Kayser M, de Knijff P : Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 2011; 12: 179–192.
Kayser M : Forensic DNA phenotyping: predicting human appearance from crime scene material for investigative purposes. Forensic Sci Int Genet 2015; 18: 33–48.
Zubakov D, Liu F, van Zelm MC et al: Estimating human age from T-cell DNA rearrangements. Curr Biol 2010; 20: R970–R971.
Zbiec-Piekarska R, Spolnicka M, Kupiec T et al: Examination of DNA methylation status of the ELOVL2 marker may be useful for human age prediction in forensic science. Forensic Sci Int Genet 2015; 14: 161–167.
Rexbye H, Petersen I, Iachina M et al: Hair loss among elderly men: etiology and impact on perceived age. J Gerontol A Biol Sci Med Sci 2005; 60: 1077–1082.
Ellis JA, Stebbing M, Harrap SB : Polymorphism of the androgen receptor gene is associated with male pattern baldness. J Invest Dermatol 2001; 116: 452–455.
Prodi DA, Pirastu N, Maninchedda G et al: EDA2R is associated with androgenetic alopecia. J Invest Dermatol 2008; 128: 2268–2270.
Brockschmidt FF, Heilmann S, Ellis JA et al: Susceptibility variants on chromosome 7p21.1 suggest HDAC9 as a new candidate gene for male-pattern baldness. Br J Dermatol 2011; 165: 1293–1302.
Hillmer AM, Brockschmidt FF, Hanneken S et al: Susceptibility variants for male-pattern baldness on chromosome 20p11. Nat Genet 2008; 40: 1279–1281.
Richards JB, Yuan X, Geller F et al: Male-pattern baldness susceptibility locus at 20p11. Nat Genet 2008; 40: 1282–1284.
Li R, Brockschmidt FF, Kiefer AK et al: Six novel susceptibility loci for early-onset androgenetic alopecia and their unexpected association with common diseases. PLoS Genet 2012; 8: e1002746.
Heilmann S, Kiefer AK, Fricker N et al: Androgenetic alopecia: identification of four genetic risk loci and evidence for the contribution of WNT signaling to its etiology. J Invest Dermatol 2013; 133: 1489–1496.
Hofman A, Darwish Murad S, van Duijn CM et al: The Rotterdam Study: 2014 objectives and design update. Eur J Epidemiol 2013; 28: 889–926.
Taylor R, Matassa J, Leavy JE, Fritschi L : Validity of self reported male balding patterns in epidemiological studies. BMC Public Health 2004; 4: 60.
Pardo LM, MacKay I, Oostra B, van Duijn CM, Aulchenko YS : The effect of genetic drift in a young genetically isolated population. Ann Hum Genet 2005; 69: 288–295.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR : MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2010; 34: 816–834.
Marchini J, Howie B, Myers S, McVean G, Donnelly P : A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007; 39: 906–913.
Akaike H : Citation classic—a new look at the statistical-model identification. CC/Eng Technol Appl Sci 1981; 51: 22–22.
Zweig MH, Campbell G : Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem 1993; 39: 561–577.
Yang J, Lee SH, Goddard ME, Visscher PM : GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ : Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015; 4: 7.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Visser M, Palstra RJ, Kayser M : Human skin color is influenced by an intergenic DNA polymorphism regulating transcription of the nearby BNC2 pigmentation gene. Hum Mol Genet 2014; 23: 5750–5762.
Visser M, Kayser M, Palstra RJ : HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res 2012; 22: 446–455.
Lee WS, Lee HJ : Characteristics of androgenetic alopecia in asian. Ann Dermatol 2012; 24: 243–252.
Hoffmann R : Male androgenetic alopecia. Clin Exp Dermatol 2002; 27: 373–382.
Birch MP, Messenger AG : Genetic factors predispose to balding and non-balding in men. Eur J Dermatol 2001; 11: 309–314.
Liu F, Wollstein A, Hysi PG et al: Digital quantification of human eye color highlights genetic association of three new loci. PLoS Genet 2010; 6: e1000934.
Liu F, van Duijn K, Vingerling JR et al: Eye color and the prediction of complex phenotypes from genotypes. Curr Biol 2009; 19: R192–R193.
Liu F, Hendriks AEJ, Ralf A et al: Common DNA variants predict tall stature in Europeans. Hum Genet 2013; 133: 587–597.
Wood AR, Esko T, Yang J et al: Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014; 46: 1173–1186.
Acknowledgements
We thank Dr David Gunn for his valuable discussions and useful comments on the manuscript. We thank Pascal Arp; Mila Jhamai; Marijn Verkerk; Lizbeth Herrera; Marjolein Peters, MSc; Carolina Medina-Gomez, MSc; and Fernando Rivadeneira, MD PhD, for their help in creating the GWAS database, and Karol Estrada, PhD; Yurii Aulchenko, PhD; and Carolina Medina-Gomez, MSc, for the creation and analysis of imputed data. We thank Sophie Flohil, Emmilia Dowlatshahi, Robert van der Leest, Leonie Jacobs, Joris Verkouteren, Ella van der Voort, and Shmaila Talib for collecting the phenotype data in the RS. This work was supported in part by the Erasmus MC University Medical Center Rotterdam and funds from the Netherlands Genomics Initiative/Netherlands Organization of Scientific Research (NWO) within the framework of the Netherlands Consortium of Healthy Ageing (NCHA).
The generation and management of GWAS genotype data for the Rotterdam Study (RS I, RS II, RS III) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands. The GWAS data sets are supported by the Netherlands Organisation of Scientific Research NWO Investments (no. 175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO), and the Netherlands Consortium for Healthy Aging (NCHA), project no. 050-060-810. FL is supported by Chinese Thousand Talent Program for Distinguished Young Scholars and MAH is supported by Unilever. The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam; Netherlands Organization for the Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); the Ministry of Education, Culture and Science; the Ministry for Health, Welfare and Sports; the European Commission (DG XII); and the Municipality of Rotterdam. The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists.
ERF Study as a part of EUROSPAN (European Special Populations Research Network) was supported by the European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007–2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the programme ‘Quality of Life and Management of the Living Resources’ of 5th Framework Programme (no. QLG2-CT-2002-01254) as well as the FP7 project EUROHEADPAIN (no. 602633). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043).
The BONN Study is supported by Heinz Nixdorf Foundation, the German Ministry of Education and Science and the German Research Council (D Glass; Project SI 236/8-1, SI236/9-1, ER 155/6-1); German Research Council (D Glass; FOR 423); and the Life and Brain GmbH (Bonn, Germany; project grant). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Liu, F., Hamer, M., Heilmann, S. et al. Prediction of male-pattern baldness from genotypes. Eur J Hum Genet 24, 895–902 (2016). https://doi.org/10.1038/ejhg.2015.220
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.220
This article is cited by
-
Genetic prediction of male pattern baldness based on large independent datasets
European Journal of Human Genetics (2023)
-
Platelet-Rich Plasma for Treatment of Hair Loss Improves Patient-Reported Quality of Life
Aesthetic Plastic Surgery (2023)
-
Biology and Criminology: Data Practices and the Creation of Anatomic and Genomic Body ‘Types’
Critical Criminology (2023)
-
Exploring the possibility of predicting human head hair greying from DNA using whole-exome and targeted NGS data
BMC Genomics (2020)
-
DNA makes an appearance
Nature Biotechnology (2018)
