
Abstract
Noonan syndrome (NS) is a genetic condition characterized by congenital heart defects, short stature and characteristic facial features. We here present the case of a girl with moderate learning disabilities, delayed language development, craniofacial features and skin anomalies reminiscent of NS. After a mutation screening of the known NS genes PTPN11, SOS1, RAF1, KRAS, GRB2, BRAF and SHOC2 we found the heterozygous c.755T>C variant in SOS1 causing the p.I252T amino-acid substitution, which was considered possibly pathogenetic by bioinformatic predictions. The same variant was present in the proband’s mother, displaying some NS features, and maternal grandfather showing no NS traits, but also by a healthy subject in 1000 genomes project database without phenotype informations. The functional analysis revealed that SOS1 c.755C activated the RAS-ERK intracellular pathway, whereas no effects on RAC-JNK cascade have been detected. After a comparison between the sequence of SOS1 cDNA from peripheral blood and SOS1 genomic DNA, we showed for the first time a differential allelic expression of the SOS1 gene in healthy individuals, thus occurring as a physiologic condition. Interestingly, we found that the mutated allele C was 50% more expressed than the wild-type allele T in all familial carriers. The comparable amount of SOS1 mRNA between mutated individuals and the controls indicates that the variant does not affect SOS1 expression. The present study provides a first evidence of allelic imbalance of SOS1 and pinpoints this condition as a possible mechanism underlying a different penetrance of some SOS1-mutated alleles in unrelated carriers.
Similar content being viewed by others


Introduction
Noonan syndrome (NS; OMIM 163950) is one of the most common Mendelian syndromes. The incidence of affected individuals is estimated to be between 1:1000 and 1:2500 with an equal male to female ratio. NS is a clinically heterogeneous disorder predominantly characterized by variable developmental delay, short stature, dysmorphic facial features, skeletal abnormalities, congenital heart defects, neck abnormalities, chest deformities and predisposition to myeloproliferative disease; mild mental retardation, bleeding diathesis, lymphedema, hearing difficulty and cryptorchidism are also occasionally observed in affected individuals.1 The diagnosis is carried out by a clinical-based scoring system, comprising major and minor criteria.2 NS is numbered among the RASopathies, a class of developmental disorders caused by germline mutations in genes encoding components or regulators of the RAS/MAPK pathway. Each RASopathy exhibits a specific clinical phenotype, but, given the involvement of a common mechanisms leading to the RAS/MAPK pathway dysregulation, they share many overlapping traits, including craniofacial dysmorphology, cardiac malformations, cutaneous and musculoskeletal abnormalities, neurocognitive impairment, hypotonia and an increased cancer risk3 with the exception of Legius syndrome.4
Compared with the other RASopathies, NS is characterized by a significant locus heterogeneity caused by the involvement of the mutated version of several RAS/MAPK pathway genes: protein tyrosine phosphatase, non-receptor type 11 (PTPN11), son of sevenless 1 (SOS1) and RAF1 in ~60–70% of NS cases, and more rarely NRAS, KRAS, BRAF, MEK, SHOC2, CBL, RIT1 and RRAS genes.1, 5, 6, 7 Both the genetics and the allelic heterogeneity can explain only in part the variable and complex NS phenotype. Furthermore, despite the identification of several NS genes, for ~30% of patients the pathogenetic mutation remains unknown and additional NS genes remain to be identified.
As far as NS genotype/phenotype correlation, mutations in the PTPN11 gene are responsible for a wide clinical spectrum, characterized by a very high incidence (of at least 80%) of congenital heart disease, mainly pulmonary valve stenosis, typical facial features, cryptorchidism and bleeding diathesis. Patients with SOS1 gene mutations display a rather distinctive form of NS with macrocephaly, ptosis, some cutaneous findings (hyperkeratotic skin, sparse eyebrows, sparse slow growing curly hair) similar to those of Cardio-facio-cutaneous syndrome and a low frequency of short stature and intellectual disability. Similarly to what observed in NS subjects with PTPN11 mutations, among individuals with SOS1 mutations cardiac defects have been reported to occur in the majority of cases (>85%), with a high prevalence of pulmonary valve stenosis, a relatively high occurrence of atrial and ventricular septal defects and a low prevalence of hypertrophic cardiomiopathy.8 RAF1 gene mutations are strongly associated with hypertrophic cardiomiopathy, intellectual disability, short stature and skin features of NS-like with lentigines syndrome (multiple lentigines, cafè-au-lait spots, multiple nevi). Patients with KRAS mutations are affected by variable intellectual disability and may manifest features overlapping those of Costello and Cardio-facio-cutaneous syndrome. Familial patients account for ~20% of NS cases and present mostly an autosomal dominant inheritance with a near-complete penetrance and variable expressivity.9 Many affected adults are diagnosed only after the birth of a more obviously affected infant, and in fact milder or subclinical phenotypes have been reported in PTPN11 and SOS1 mutation carriers.10, 11 The reported different degrees of severity of NS phenotype in both familial and sporadic carriers, for the same or similar functional mutations, are suggestive of the involvement of cis/tr ans regulatory factors, modulating the effects of specific mutations that should be investigated.
We here demonstrate the hyperactivating function of the SOS1 c.755T>C variant occurring in a NS family, and for the first time we show a differential allelic expression (DAE) of SOS1 gene that might have a role in both the complete penetrance and the variable expressivity of the NS clinical phenotype.
Materials and methods
Mutation screening and sequencing of GC-rich stretches
Peripheral lymphocyte DNA was obtained from patient, parents, maternal grandparents and controls using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. Informed consent for molecular genetic studies on DNA and RNA was signed by all the analyzed patients. Sequencing was performed on the coding region of PTPN11 (NG_007459.1), the nine exons of SOS1 (NG_007530.1), and three exons of RAF1 (NG_007467.1) (exons 7, 14 and 17), where mutations have been previously identified,10, 12, 13 KRAS (NG_007524.1), GRB2 (NG_029556.1), exons 6–11–12–13–15 of BRAF (NG_007873.3) and exon 2 of SHOC2 (NG_028922.1). Primer sequences are published elsewhere.10, 11 PCR was carried out in a 50 μl reaction volume containing 150 ng of genomic DNA, 0.2 μ M primers, 100 μ M dNTPs, 10 × reaction buffer, 50 mM MgCl2 and 2.5 U GoTaq DNA Polymerase (Promega, Madison, WI, USA) with the following cycling profile: 4 min initial denaturation at 95 °C, 35 cycles as follows: 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s.
The primer sequences for the amplification of the SOS1 promoter region and for the SOS1 3′-UTR regions are reported in Supplementary Table S1. The GC-rich stretch, located in the SOS1 promoter region, was divided into four fragments and amplified using AccuPrime GC-Rich DNA Polymerase (Life Technologies, Carlsbad, CA, USA). The PCR was carried out in a 25 μl reaction volume containing 100 ng of genomic DNA and 10 μ M primers, following the manufacturer’s instructions. The fragments were amplified with the following cycling profile: 5 min initial denaturation at 95 °C, 30 cycles as follows: 95 °C for 30 s, 54 °C for 30 s, 72 °C for 1 min. The PCRs for the SOS1 3′-UTR regions were carried out using GoTaq DNA Polymerase (Promega) in a 25 μl reaction volume containing 100 ng of genomic DNA, 10 μ M primers, 100 μ M dNTPs, 10 × reaction buffer and 50 mM MgCl2. The following cycling profile was carried on: 4 min initial denaturation at 95 °C, 30 cycles as follows: 95 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s. The specificity of all the amplified PCR products was checked by 1.5% agarose gel electrophoresis. PCR products were bi-directionally sequenced using the Terminator v3.1 Cycle Sequencing Kit (Life Technologies), and resolved on an automated ABI-3130xl DNA genetic analyzer (Life Technologies). Output data were analyzed by SeqScape software v2.5 (Life Technologies).
The data concerning the variant and the phenotype of the proband have been submitted to the public database LOVD (https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php?select_db=SOS1): the patient ID is 59 and the variant accession number is SOS1_00027.
Plasmids and mutagenesis
Plasmid hSOS1 (human Sos WT) was a kind gift of G Scita (IFOM, Milano); hSOS1, hSOS1-M269R and hSOS1-R497Q were previously described.11 The hSOS1 plasmid was used as template for site-directed mutagenesis, performed using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) to create the hSOS1-I252T mutated plasmid. Pairs of complementary mutagenic primers (I252T-Fw: 5′-TCGCATAGTAGATACACATGAACTTAGTG-3′ and I252T-Rev: 5′-CACTAAGTTCATGTGTATCTACTATGCGA-3′) were designed. Hemagglutinin (HA)-c-Jun NH2-terminal kinase 1 (JNK1) and HA-tagged extracellular regulated kinase1 (HA-ERK1) have been previously described.14
Biochemical analysis
Human embryonic kidney HEK293 cells were grown in DMEM plus 10% FBS and antibiotics. Cells were transiently transfected with equal amounts of the full-length hSOS1 or the mutated isoforms of hSOS1 and either HA-tagged JNK1 or ERK1 using the JetPEI (Polyplus, New York, NY, USA) method according to the manufacturer’s instructions. Twenty-four hours after transfection the medium was replaced with serum-deprived medium and left for 18 h. HA-ERK1- and HA-JNK1- transfected cells were then stimulated with epidermal growth factor (EGF) (20 ng/ml) for 10 and 15 min, respectively. Cells were then collected in lysis buffer (Tris–HCl 50 mM, pH 7.5, NaCl 150 mM, Triton X-100 1%, glycerol 10%, NaF 50 mM, NaVO3 1 mM and cocktail of proteases inhibitors) and equal amounts of proteins were immunoprecipitated using anti-HA antibodies (BABCO, Richmond, CA, USA) and subsequently incubated with protein G slurry. Beads were washed and resuspended in SDS sample buffer. Samples were separated on SDS–PAGE and then transferred to nitrocellulose membranes. The membranes were then incubated overnight at 4 °C with the primary antibodies: anti-HA (1:1000), anti-phospho ERK1/2 (1:1000, Cell Signalling, Danvers, MA, USA), anti-phospho-JNK1 (1:500, Cell Signalling) or anti-SOS1 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). After the incubation with the horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Little Chalfont, UK), the membranes were developed using ECL plus kit (GE Healthcare), according to the manufacturer’s instructions. Band intensities were then quantified by densitometry using ImageJ software (National Institute of Health, USA).
RNA extraction and qPCR
RNA extraction from blood of patients and controls was performed using the Tempus Blood RNA tubes and the Tempus Spin RNA Isolation kit (Life Technologies).
Total RNA (1 μg) was reverse-transcribed using the High Capacity cDNA Reverse Transcription kit (Life Technologies). PCR was performed using 1 μg of cDNA and specific primers for SOS1 transcript (NM_005633.3) Sos1-ex6-fw: 5′-GCAGACATTTTAAAGCTGGTTGG-3′ and Sos1-ex6-rev: 5′-CTCTGCTAAGTCTTCAAAGCAG-3′ for the amplification of the c.755T>C region or Sos1-3utr-fw: 5′-TTCACCATCTAAGATTATGT-3′ and Sos1-3utr-rev: 5′-CATTTTGTTTGGGTGTGTGG-3′ for the amplification of the rs1059310:T>C SNP region using GoTaq DNA polymerase (Promega) and direct sequenced as described above. The pick height in electropherograms derived from direct sequencing was measured using ChromasPro software. Statistical analysis was performed using a student’s t-test.
qPCR was performed using GoTaq qPCR master mix (Promega) and primers qPCR-Sos1-fw: 5′-TCCACGAAGACGACCAGAAT-3′ and qPCR-Sos1-rev: 5′-GGGACTGTCCAAATGCTTA-3′. GAPDH was amplified as an internal control.
Results
Case report
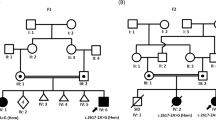
A 9 years and 5-month-old female (patient III-2, Figure 1) born to healthy unrelated parents was referred for genetic assessments because of moderate learning disabilities. Family history was noncontributory.

Pedigree with photographs and summary of clinical features of NS family members bearing the c.755T>C variant in SOS1. Proband (III-2), shown by the arrow, and her mother are colored in black to indicate that they display the full NS phenotype. Her maternal grandfather is colored in gray to indicate a milder phenotype (see also Table 1).
At the time of her birth, her mother was 31 and her father 32-year-old. Pregnancy was not problematic. She was born after 40 weeks of gestation with weight of 3070 g (25th centile), a length of 50 cm (50th centile) and occipitofrontal circumference of 35 cm (75–90th centile). Delivery was carried out after a prolonged labor with cesarean section because of a previous cesarean section. Apgar score was 4 and 10 at 1 and 5 min. She walked unassisted at 12 months and was toilet trained at 30 months. She was able to say her first words at 10 months; however at kindergarten her speech was not easily understandable and her language was still composed of simple sentences at almost 10-year-old. Neuropsychological evaluation showed borderline cognitive skills. At the time of clinical evaluation at 9 years and 5 months, a myopathic appearance of the face with bilateral ptosis was evident, together with strabismus, a high forehead, micrognatia and slight webbing (Figure 1). Face was asymmetric, the left side being smaller. Cutis marmorata was more evident at the lower limbs than at the upper limbs. Abnormalities of skin pigmentation included cafe-au-lait spots, lentigines and vitiligo areas at the anterior face of both legs, sternum, groin and right scapula. First manifestations of vitiligo were evident on the legs at 7 years. Occipitofrontal circumference was 53 cm (75th centile), weight 25 Kg and height 127 cm (between the 10th and 25th centile). Hypotonia was appreciable at the upper limbs. Brain MRI scan images, carried out when the patient was 9 years and 10-months-old, showed a small arachnoid cyst in the left temporal lobe. EEG was normal; antinuclear antibodies were detected as the result of autoimmune diseases screening carried out for vitiligo. Ptosis, relative macrocephaly, webbing, borderline cognitive skills, cafe-au-lait spots, lentigines, hypotonia and vitiligo as possible sign of an autoimmune disorder suggested to investigate the spectrum of the Neuro-cardio-facio-cutaneous syndrome, especially NS. Heart and kidneys ultrasound carried out on the basis of the diagnostic hypothesis were normal. Karyotype was 46,XX; fragile X syndrome was ruled out through DNA evaluation. Clinical genetics examination of the probands’parents and maternal grandparents was carried out subsequently the SOS1 testing. Proband’s mother (II-1) was evaluated at 41 years. Figure 1 shows her thick eyebrows, bilateral ptosis, broad and prominent nasal root, short neck, her ears are mildly low-set and posteriorly rotated. Skin examination revealed four cafe-au-lait spots in subscapulary region and a number of pigmented naevi on chest and arms. Skin creases pattern over proximal interphalangeal joints of both hands was simplified. Hypotonia was appreciable at the upper limbs. Occipitofrontal circumference was 57 cm (>97th centile) and height 164 cm (>50th). Developmental and/or learning disabilities were denied; she appeared indeed a very appropriate person. As for the proband’s maternal grandfather (I-2), at the time of clinical evaluation at 71 years (Figure 1), there were no relevant dysmorphic features except high-arched eyebrows. Occipitofrontal circumference was 59.5 cm (>97th centile), height 166.5 cm (10th). There were lentigines at the upper limbs and hands. Phenotype of the proband’s father (II-2) and maternal grandmother (I-1) was unremarkable.
Mutation analysis
Patient III-2 was subjected to a mutation screening of the known NS genes PTPN11, RAF1, SOS1, KRAS, SHOC2, BRAF and also GRB2, which was considered as a candidate gene in a previous study.11 We found the heterozygous variation c.755T>C in exon 6 of SOS1 (NG_007530.1), leading to the p.(I252T) amino-acid substitution. The same variant was present in the mother and maternal grandfather, both displaying some clinical NS features (Table 1). SOS1 encodes for a 150 kDa Ras-GEF (guanine exchange factor) protein characterized by a RAS-GEF (Cdc25) domain, conserved histone-like fold, Dbl homology (DH) and pleckstrin homology (PH) domains, a helical linker, a RAS exchange motif (REM) and a proline-rich region. SOS1 is also known to activate a distinct pathway mediated by RAC1, a GTPase belonging to the Rho-family.15 Ile252 is an invariant residue in a hydrophobic core of the SOS1 DH domain that, competing with RAS for binding to the allosteric site, might act as an intramolecular inhibitor of RAS-GEF activity.16 Bioinformatic analysis with PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) identified the p.(I252T) variant as ‘probably damaging’ with a score of 0.674 and the analysis with SIFT predicted the amino-acid change to ‘affect protein function’, given that the Ile residue is highly conserved during evolution.
The c.755T>C variation has been already reported in a NS familial case, but no validation studies have been carried out.8 A more recent NMR-based study, despite detecting a functional effect of p.(I252T) variant in solution, failed to show ERK activation in serum-activated cells.17 Furthermore, this variant is present in the SNP databases (dbSNP database http://www.ncbi.nlm.nih.gov/SNP/ rs142094234:T>C, chr2.hg19:g.39051253A>G) with a frequency <0.001, being observed only 1 time out of 1000 genomes (http://www.1000genomes.org). For these reasons we wanted to check a possible pathological effect of this variant.
Functional study of SOS1 c.755T>C (p.(I252T))
To investigate whether the SOS1 c.755T>C (p.(I252T)) variant could lead to a functional variation of the Ras pathway activity, we verified whether it hyperactivates the ERK1 downstream effector. At this purpose we transfected HEK293 cells with the mutant Sos-p.I252T plasmid, the wild-type human SOS1 or the mutant isoform SOS1 c.806T > C (p.(M269R)), previously reported to activate the RAS-ERK pathway13 as a positive control and compared the ERK1 phosphorylation levels of each SOS1 isoform before and after EGF stimulation. We found that the Sos-p.I252T substitution significantly increased ERK1 phosphorylation level, with a level comparable to the c.806T > C (p.(M269R)) mutant-positive control (Figure 2a and b).

ERK and JNK activation assays. (a) hSOS1 (Sos WT) or M269R or I252T together with HA-ERK1 were expressed in HEK293 cells. Cells were serum starved for 18 h and then stimulated with EGF (20 ng/ml) for 10 min. HA-ERK1 was immunoprecipitated with anti-HA antibodies and the level of phosphorylated ERK1 (upper row) and HA-ERK1 (middle row) in the immunoprecipitates was detected with anti-phospho-ERK and anti-HA antibodies, respectively. The level of the different SOS1 constructs was detected in total cell extracts with antibodies against SOS1 (bottom row). (b) The graph represents the mean±SEM of phospho-ERK1 induction normalized to HA-ERK1 intensity in the immunoprecipitates. The quantification was carried out on three independent experiments, and the data are expressed as the ratio normalized to the unstimulated condition. Mann–Whitney test has revealed a significant increase of ERK1 phosphorylation upon EGF stimulation in comparison with the not-stimulated condition for each SOS1 construct (Sos WT+EGF vs Sos WT not stimulated *P<0.05, M269R+EGF vs M269R not stimulated **P<0.01, I252T+EGF vs I252T not stimulated ***P<0.001). (c) hSOS1 (Sos WT) or R497Q or I252T together with HA-ERK1 were expressed in HEK293 cells. Cells were serum starved for 18 h and then stimulated with EGF (20 ng/ml) for 15 min. HA-JNK1 was immunoprecipitated with anti-HA antibodies and the level of phosphorylated JNK1 (upper row) and HA-JNK1 (middle row) in the immunoprecipitates was detected with phospho-JNK and HA antibodies, respectively. The level of the different SOS1 constructs was detected in total cell extracts with antibodies against SOS1 (bottom row). (d) The graph represents the mean±SEM of phospho-JNK1 induction normalized to HA-JNK1 intensity in the immunoprecipitates. The quantification was carried out on three independent experiments, and the data are expressed as the ratio normalized to the unstimulated condition for each SOS1 construct. Mann–Whitney test has shown that EGF stimulation significantly increases the level of JNK phosphorylation in SOS WT and in R497Q-transfected cells in comparison with the not-stimulated condition (Sos WT+EGF vs Sos WT not stimulated *P<0.05, R497Q+EGF vs R497Q not stimulated *P<0.05).
As SOS1 is also involved in RAC1 pathway signal transduction regulation, we sought to determine whether this pathway might be affected by detection of variations in JNK activation. HEK293 cells have been transfected, under EGF stimulation, with the mutant p.I252T, the human wild-type SOS1 and the mutant c.1490G>A (p.(R497Q)), previously reported to activate the RAC-JNK pathway,11 as a positive control. After HEK293 cells transfection with the Sos-p.I252T plasmid, JNK was not found to be significantly overactivated under EGF stimulation, in comparison with the Sos-wt or the JNK activating SOS1 c.1490G>A (p.(R497Q)) mutant (Figure 2c and d).
DAE analysis of SOS1 gene
Because the c.755T>C variant has also been detected in a healthy individual, being reported in the 1000 genomes SNP database, we hypothesized that SOS1 alleles may be differentially expressed as well-affecting/unaffecting variants sporadically occurring in population, as described for other genes,18 and that this mechanism may have a role in the penetrance of potential pathogenetic variants. As SOS1 allelic expression was never investigated, we studied the expression of the common SNP rs1059310:A>G, localized within the SOS1 3′-UTR, in five heterozygous healthy controls. The SOS1 rs1059310:A>G SNP was PCR amplified from both genomic DNA and peripheral blood cDNA and subjected to direct sequencing. The heights of the A and G picks in the resulting electropherograms were evaluated and the A:G allele ratio of both genomic DNA and cDNA was compared. The A:G allele ratio was about 50:50 in genomic DNA as expected but was significantly different (A:G allele ratio mean 62:38) in cDNA samples (Figure 3a).

SOS1 allelic expression analysis. (a) A:G allele ratio of the common rs1059310:A>G SNP in genomic DNA and cDNA of five controls (student's t-test **P<0.005). (b) Representative electropherograms of PCR amplified fragments from genomic DNA or cDNA of individuals heterozygotic for the SOS1 c.755T>C variant (boxed). (c) The height of the picks in the above electropherogram has been measured and the C:T allele ratio calculated for genomic DNA and cDNA. Statistical analysis was performed on five independent PCR amplifications. The genomic bar represent the mean±SD of the allele ration of all the subjects (student's t-test **P<0.005 compared with genomic). (d) qPCR of SOS1 mRNA after retrotranscription of total RNA extracted from blood samples of mutated family members and controls. The control group includes the maternal grandmother (I-1) of patient III-2. GAPDH was used in qPCR as an internal normalizer. The histogram shows the means±SD of three independent experiments.
To verify whether the SOS1 c.755T>C alleles also showed DAE, a fragment of SOS1 exon 6 including the c.755T>C variant was PCR amplified from both genomic DNA and peripheral blood cDNA of family carriers and subjected to direct sequencing. Sequence analysis of both genomic DNA and cDNA allowed us to establish that the C:T allele ratio in the cDNA of all familial carriers was 62:38, indicating that the mutated allele C was expressed 1.6-folds more than the wild-type allele T (Figure 3b and c). The comparable ratio value of mRNA level of SOS1 alleles in both NS family members and healthy subjects indicates that SOS1 DAE can normally occur.
To test whether the higher expression of the mutated allele could result in an increased amount of the total SOS1 mRNA in our NS patients, we performed a qPCR on total RNA from the NS family members and the previously described controls. The total amount of SOS1 mRNA was comparable between mutated individuals and controls, which included also the non-mutated healthy grandmother (I-1) of the proband (Figure 3d).
Search for SNPs’ heterozygosity of SOS1 promoter region and miR-124-binding site in NS patients and controls showing DAE
Starting from the central assumption that in the absence of cis-regulatory polymorphisms, both copies of an autosomal gene will contribute equal amounts of messenger RNA, we searched for SNPs’ heterozygousity at cis-regulatory sites responsible for SOS1 transcription, mRNA stability and/or translational efficiency in all the three NS family members here described and in the five controls showing DAE. We sequenced the SOS1 promoter region (chr2.hg19:g.39,347,347_39,348,479 UCSC database) and the 3′UTR validated miR-124-binding site into which the SNP rs115465194:A>G is located.19 We observed a homozigosity condition of the most frequent allele for each of the eighteen SNPs analyzed (1000 genomes database http://www.1000genomes, Supplementary Table S2).
Discussion
We here report on the functional study of SOS1 c.755T>C (p.(I252T)) variation that we found in a NS family from Northern Italy. The c.755T>C variant is located in exon 6 within the SOS1 DH domain and it has so far been identified only in another NS patient.8 As already observed by Lepri et al,8 we predicted that the variant might lead to a structural perturbation of the DH fold, destabilizing the masking of the distal RAS-binding site. The SOS1 DH domain interacting with the PH domain both inhibits exchange activity for RAC1 and promotes the interaction with the cell membrane inhibiting RAS activation.15 As the SOS1 c.755T>C variation has been recently reported also in a healthy subject (http://www.1000genomes.org), its role in activating the RAS pathway needed to be demonstrated. Furthermore, a study based on NMR methodology aimed at detecting defects in RAS GTPase cycling showed that p.I252T variant failed to stimulate ERK activation in serum-activated cells, whereas the assays carried out in solution allowed to detect ERK hyperactivation. This apparent contradictory result was explained by the layered interdomain interactions and association with cell membranes that would prevent the hyperactivation effect of this variant.17 We here show that the SOS1 c.755T>C (p.(I252T)) variant detected in our NS family hyperactivates the RAS pathway. Accordingly, several other missense mutations have been identified in the DH domain of SOS1 (Tartaglia et al1) and in particular the c.797C>A (p.(T266K)) and c.806T > C (p.(M269R)) changes in exon 6 have been shown to be pathogenetic mutations.13
The three affected members of the NS family here described did not show any mutation in the known NS genes other than the c.755T>C SOS1 variant. Our proband’s phenotype resembles that of SOS1-mutated individuals, being associated with relative macrocephaly, ptosis and absence of short stature, whereas her cutaneous findings – lentigines, cafè-au-lait spots – are similar to those of NS-like with lentigines rather than those found in Cardio-facio-cutaneous syndrome and patients with SOS1 gene mutations,8 that is, hyperkeratotic skin, sparse eyebrows, sparse slow growing curly hair. Our proband IQ was borderline to the lower limit of the normal range and displayed impairment of language and speech development and of learning at school, in agreement with the observation of a low frequency (11%) of intellectual disability in SOS1-mutated patients that, if present, usually causes only minor impairment in affected individuals.8 The SOS1 c.755T>C variant was also found in the proband’s mother and in her maternal grandfather. As detailed in Table 1, both the proband and her mother fulfill the diagnostic criteria for a definite NS if family history is accounted for. The maternal grandfather, displaying only macrocephaly, short and broad neck, high-arched eyebrows, freckles on hands and forearms, has a phenotype less suggestive for NS, even taking into account that increasing age makes characteristic facies more subtle. The previously reported NS patient bearing the SOS1 c.755T>C variant8 was familial as well and belonged to a cohort of subjects with features fitting NS, or highly suggestive of NS, but, because the phenotype was not further detailed within the cohort, comparison between the two SOS1 c.755T>C-mutated families cannot be performed. Despite the apparent genetic homogeneity, the proband and her mother show the full NS phenotype, whereas the grandfather has features that seem mixed up with those of the general population. An incomplete expression of the full NS phenotype with subclinical traits has already been reported in some SOS1 mutation carriers.10, 11 Interestingly, the inherited c.1490G>A (p.(R497Q)) SOS1 mutation, activating Rac1 pathway, has been detected together with a validated pathogenetic de novo RAF1 mutation in the same patient, showing the possibility of the co-expression of different mutant effectors of the RAS pathway, leading to the onset of variable NS phenotypes.11 Similarly to what occurs in the family here described, in the SOS1 c.1490G>A (p.(R497Q)) mutation family there was a fading in the craniofacial phenotype when dating back to the third generation, but they all showed keratosis pylaris, a SOS1 distinctive ectodermal abnormality.8, 10 The functional study here reported, showing a strong activation of RAS-ERK signalling following EGF treatment that, not detected for RAC-JNK, excludes the direct hyperactivation of the Rac1 pathway, consistently with the association between skin involvement and SOS1 c.1490G>A (p.(R497Q)) mutation. The presence of further NS-specific signs as a mild thorax deformity and CHD (pulmonary valve stenosis, hypertrophic cardiomyopathy) in the proband was interpreted as the result of the occurrence of the RAF1 mutation.11 Therefore, it is possible that specific isoforms of modifier genes might modulate the onset of some NS traits in affected individuals of the same family. Alternatively, considering the mild expression of the proband’s phenotype even if fitting the full NS, the different genetic background and/or the different environmental influences could have contributed to decrease the expression of the c.755T>C SOS1 variant across the family generations, that is, with the degree of relationship.
Another possible explanation of the clinical heterogeneous expressivity despite homogeneity of DAE in the three family members could come from a tissue-specific allelic imbalance or a tissue differential sensitivity to the genetic variation: for example, differently compared with her family members, the proband’s brain may ‘behave’ like blood tissue.
The detection of the SOS1 c.755T>C also in a healthy subject (http://www.1000genomes.org), even if with a very low frequency (<0.001), prompted us to hypothesize that epigenetic mechanisms might influence the penetrance and/or variable expression of SOS1 c.755T>C pathogenetic allele and therefore enhancing or decreasing the activating effect of this variant. We first investigated and verified the occurrence of DAE. The patterns of allelic imbalance represent a common biological phenomenon affecting human gene expression. It was estimated that in lymphoblastoid cell lines ~20% of genes display population-average ratios of allelic expression larger than 40:60 or an higher than expected variance in allelic expression among heterozygous individuals.18 This condition might reflect the presence of different, possibly unknown, causative variants interfering with the complex modulation of gene expression.20, 21 Accordingly, analyzing the common rs1059310:A>G SOS1 SNP in five healthy heterozygous individuals, we verified the occurrence of SOS1 DAE. Because the rs1059310 A allele resulted more expressed than the G allele in all analyzed individuals (60:40 ratio), we speculate that it might be in linkage disequilibrium with a putative gene expression regulatory motif. As far as the c.755T>C variant, our family members showed a higher expression of the mutated allele in respect to the wild type, possibly increasing the activating effect of the variant. The fact that the mutated allele did not increase the total SOS1 mRNA level, but resulted in an allelic expression imbalance, suggests that the c.755T>C variation does not affect SOS1 expression. We speculated that this variant occurs, in this family, in cis with a putative SOS1 SNP, increasing the level of mutated mRNA, or alternatively, in trans with a SNP leading to a decrease of the normal mRNA, resulting in a higher level of the SOS1 protein able to increase the activation of the Ras downstream Erk1 effector. We searched for the heterozygosity of SNPs located in regulatory regions, mainly in the promoter and in proximity to the validated miR-124 binding site in the 3′UTR,19 but we did not detect this condition in the NS family members. It has been estimated that in over 40% of all genes individual in human population at functional cis-regulatory sites is heterozygous22, 23 and DAE affects from 20 to 50% of genes.18 These functional variants remain difficult to identify as regulatory regions can reside hundreds of thousands of bases away from the transcriptional start site.24, 25
We propose that DAE can explain the lack of NS phenotype in the control despite carrying the above variant, which probably occurred in trans with a putative SNP increasing the allele expression and that variations in the expression of the normal or mutated SOS1 allele may be involved in the expression intensity of the NS phenotypes, which in some cases differs among unrelated individuals affected with the same mutations, as it has been already proposed for neurofibromatosis type 1.26 However, as no clinical information is available about the control individual carrying the c.755T>C SOS1 variant, a very mild NS phenotype, similar to our proband’s mother, cannot be ruled out.
In conclusion, the obtained results demonstrated the pathogenic role of SOS1 c.755T>C variant. The present study provides the first evidence of DAE for SOS1 gene in both NS and healthy individuals, suggesting that DAE is a physiological condition during SOS1 gene expression. Moreover, it adds for the first time the evidence that DAE, at least analyzed in blood tissue, is not sufficient to explain the reduced penetrance and/or expressivity of the NS phenotype across family generations, which is a well-known phenomenon.
SOS1 mutations account for 10–13% of NS patients13 and probably in some cases, when a certain mutated allele is underexpressed, although functionally activating, it is not so penetrant to cause the disease. The coupling of both SOS1 sequencing and the DAE analyses might be recommended to evaluate the pathogenetic potential of the identified SOS1 variant suggesting, when the mutated mRNA is underrepresented, sequence analysis of additional NS genes.
The findings here reported pinpoint on the role of possible epigenetic mechanisms leading to DAE, which might emerge as modifier factors able to modulate the phenotypic effects of a specific mutation.
References
Tartaglia M, Zampino G, Gelb BD : Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol 2010; 1: 2–26.
van der Burgt I, Berends E, Lommen E, van Beersum S, Hamel B, Mariman E : Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet 1994; 53: 187–191.
Rauen KA : The RASopayhies. Annu Rev Genomics Hum Genet 2013; 14: 355–369.
Brems H, Pasmant E, Van Minkelen R et al: Review and update of SPRED1 mutations causing Legius syndrome. Hum Mutat 2012; 33: 1538–1546.
Martinelli S, De Luca A, Stellacci E et al: Heterozygous germline mutations in the CBL tumor supressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet 2010; 15: 250–257.
Aoki Y, Niihori T, Banjo T et al: Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet 2013; 93: 173–180.
Flex E, Jaiswal M, Pantaleoni F et al: Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet 2014; 23: 4315–4327.
Lepri F, De Luca A, Stella L et al: SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat 2011; 32: 760–772.
Tartaglia M, Gelb BD : Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet 2005; 6: 45–68.
Tartaglia M, Pennacchio LA, Zhao C et al: Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 2007; 39: 75–79.
Longoni M, Moncini S, Cisternino M et al: Noonan syndrome associated with both a new Jnk-activating familial SOS1 and a de novo RAF1 mutations. Am J Med Genet A 2010; 152A: 2176–2184.
Pandit B, Sarkozy A, Pennacchio LA et al: Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 2007; 39: 1007–1012.
Roberts AE, Araki T, Swanson KD et al: Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 2007; 39: 70–74.
Innocenti M, Zippel R, Brambilla R, Sturani E : CDC25(Mm)/Ras-GRF1 regulates both Ras and Rac signaling pathways. FEBS Lett 1999; 460: 357–362.
Pierre S, Bats AS, Coumoul X : Understanding SOS (Son of Sevenless). Biochem Pharmacol 2011; 82: 1049–1056.
Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J : Structural analysis of autoinhibition in the Ras activator Son of Sevenless. Cell 2004; 119: 393–405.
Smith MJ, Neel BG, Ikura M : NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A 2013; 110: 4574–4579.
Serre D, Gurd S, Ge B et al: Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet 2008; 4: e1000006.
Lv Z, Yang L : MiR-124 inhibits the growth of glioblastoma through the downregulation of SOS1. Mol Med Rep 2013; 8: 345–349.
Wilkins JM, Southam L, Price AJ, Mustafa Z, Carr A, Loughlin J : Extreme context specificity in differential allelic expression. Hum Mol Genet 2007; 16: 537–546.
Burkhardt J, Kirsten H, Wolfram G, Quente E, Ahnert P : Differential allelic expression of IL13 and CSF2 genes associated with asthma. Genet Mol Biol 2012; 35: 567–574.
Ayala FJ, Escalante A, O’Huigin C, Klein J : Molecular genetics of speciation and human origins. Proc Natl Acad Sci USA 1994; 91: 6787–6794.
Rockman MV, Wray GA : Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol 2002; 19: 1991–2004.
Kleinjan DA, van Heyningen V : Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 2005; 76: 8–32.
Liu J, Francke U : Identification of cis-regulatory elements for MECP2 expression. Hum Mol Genet 2006; 15: 1769–1782.
Jentarra GM, Rice SG, Olfers S, Rajan C, Saffen DM, Narayanan V : Skewed allele-specific expression of the NF1 gene in normal subjects: a possible mechanism for phenotypic variability in neurofibromatosis type 1. J Child Neurol 2012; 27: 695–702.
Acknowledgements
We thank Dr Milena Crippa and Dr Valeria Rimoldi for their technical support. This study was supported by an academic fund for basic research (Programma dell’Università per la Ricerca PUR to PR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Moncini, S., Bonati, M., Morella, I. et al. Differential allelic expression of SOS1 and hyperexpression of the activating SOS1 c.755C variant in a Noonan syndrome family. Eur J Hum Genet 23, 1531–1537 (2015). https://doi.org/10.1038/ejhg.2015.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.20
