
Abstract
Mutations in the Aristaless-related homeobox gene (ARX) lead to a range of X-linked intellectual disability phenotypes, with truncating variants generally resulting in severe X-linked lissencephaly with ambiguous genitalia (XLAG), and polyalanine expansions and missense variants resulting in infantile spasms. We report two male patients with early-onset infantile spasms in whom a novel c.34G>T (p.(E12*)) variant was identified in the ARX gene. A similar variant c.81C>G (p.(Y27*)), has previously been described in two affected cousins with early-onset infantile spasms, leading to reinitiation of ARX mRNA translation resulting in an N-terminal truncated protein. We show that the novel c.34G>T (p.(E12*)) variant also reinitiated mRNA translation at the next AUG codon (c.121–123 (p.M41)), producing the same N-terminally truncated protein. The production of both of these truncated proteins was demonstrated to be at markedly reduced levels using in vitro cell assays. Using luciferase reporter assays, we demonstrate that transcriptional repression capacity of ARX was diminished by both the loss of the N-terminal corepressor octapeptide domain, as a consequence of truncation, and the marked reduction in mutant protein expression. Our study indicates that premature termination mutations very early in ARX lead to reinitiation of translation to produce N-terminally truncated protein at markedly reduced levels of expression. We conclude that even low levels of N-terminally truncated ARX is sufficient to improve the patient’s phenotype compared with the severe phenotype of XLAG that includes malformations of the brain and genitalia normally seen in complete loss-of-function mutations in ARX.
Similar content being viewed by others

Introduction
Aristaless-related homeobox gene (ARX) (NM_139058.2) (MIM: 300382) is a paired-type homeodomain transcription factor with critical roles in development.1 Mutations in ARX cause X-linked intellectual disability (ID) with over 44 different mutations in ARX found in over 100 families, including some sporadic cases.1 ID is a consistent feature, with some mutations in ARX leading to a broad spectrum of seizure phenotypes, including X-linked infantile spasms (ISSX), West syndrome, infantile epileptic-dyskinetic encephalopathy, and early infantile epileptic encephalopathy (also known as Ohtahara syndrome).1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 Other mutations in ARX lead to severe malformations of the brain and genitalia such as Proud syndrome with agenesis of the corpus callosum and abnormal genitalia,20 hydranencephaly with abnormal genitalia,20, 21 and X-linked lissencephaly with abnormal genitalia (XLAG).1, 20, 21, 22, 23, 24, 25, 26, 27, 28 Mutations that lead to severe malformation phenotypes are usually protein truncation and point mutations in critical residues, leading to complete loss-of-function ARX protein.
We report a novel truncating mutation in ARX in a family with two affected brothers with early-onset infantile spasms. The mutation identified in the proband, c.34G>T (p.(E12*)), is predicted to yield a severely truncated ARX protein only 11 amino acids in length (p.M1_S11), and thus give rise to a non-functional or degraded ARX protein. However this patient had no malformations consistent with a complete loss of ARX function, and his brother had an even milder phenotype. Previously, a c.81C>G (p.(Y27*)) variant in ARX was identified in two male cousins with early-onset infantile spams and was shown to be able to reinitiate mRNA translation at the next AUG (p.M41) codon and escape nonsense-mediated decay (NMD), producing an N-terminally truncated ARX protein (p.M41_C562).2 In this study, we show that the c.34G>T (p.(E12*)) ARX mutation identified in this family also reinitiates mRNA translation at the next AUG codon (c.121–123 (p.M41)), producing the same N-terminally truncated ARX protein (p.M41_C562) missing the octapeptide domain. We demonstrate that these N-terminally truncated proteins were expressed at a markedly reduced level. Our analysis indicates that compromised transcriptional repression capacity of ARX is likely to result from the marked reduction in protein abundance as much as the loss of the N-terminal octapeptide corepressor domain. Although our patients do have early-onset infantile spasms, we suggest that even quite low levels of N-terminally truncated protein results in a less severe outcome compared with the very severe malformation phenotype of XLAG. Our data suggests that even small improvements in the abundance of this critical transcription factor may alleviate the severity of the clinical phenotype.
Materials and methods
Patients
The proband (ClinVar database ID SCV000192096) was referred to the epilepsy center at Nationwide Children’s Hospital, Ohio State University (Columbus, OH, USA) at 7 months of age for intermittent jerking movements involving the head and extremities. The child’s past medical history was significant for delay in achieving developmental milestones, congenital torticollis, plagiocephaly, strabismus, visual impairment (difficulty fixating on objects), and irritability of recent onset. The neurological evaluation revealed a hypotonic infant with significant head lag, plagiocephaly, and limited visual tracking ability. He had mild dysmorphism consisting of anterior rotation of the right pinna, mild hypertelorism, and high-arched palate. He could not sit without support, and deep tendon reflexes were difficult to elicit. Brain MRI only had evidence of tortuosity of the intraorbital segment of the optic nerve bilaterally (Figures 1a and b). Video electroencephalogram (EEG) confirmed the suspicion of infantile spasms. The background showed hypsarrhythmia with high amplitude, posteriorly predominant multifocal spikes, and polyspike-wave discharges. During sleep, the spikes had a ‘grouping tendency’ creating a burst suppression-like pattern (Figures 1c and d). The clinical events consisted of clusters of flexor spasms involving all extremities, with associated upward eye deviation. Ictal EEG consisted of short ‘electrodecrements’ preceded by generalized spike-wave discharges with posterior predominance or fast posterior polyspikes (Figure 1e). Basic metabolic panel and chromosomal microarray analysis were reported to be normal.

Magnetic resonance imaging (MRI) brain scan and EEG before and after ACTH treatment of patient. (a) Sagittal T1 MRI of the brain showing normal gyral pattern and (b) axial T2 MRI of the brain showing tortuosity of the intracranial portion of the optic nerves (arrow). (c–e) Before ACTH treatment. (c) EEG during wakefulness. Record showed hypsarrhythmia with high amplitude multifocal spikes. (d) EEG during sleep. High amplitude spikes with a ‘grouping tendency’ distorting the sleep architecture (depicted by brackets). (e) Ictal EEG. ‘Electrodecremetal response’ associated with clinical spasm (depicted by bracket). (f–g) Two weeks after ACTH treatment. (f) EEG during wakefulness. Record showed resolution of hypsarrhythmia. The occipital spike-wave discharges were still present (depicted by bracket). (g) EEG during sleep. The sleep EEG showed resolution of hypsarrhythmia. Epileptiform discharges were present in the posterior quadrants (depicted by brackets). (h–i) EEG at 33 months of age. (h) EEG during wakefulness. Background during wakefulness showed multifocal discharges with posterior predominance (depicted by brackets). (i) EEG during sleep. Bursts of diffuse spike-wave discharges and multifocal spikes (depicted by brackets). All traces in (c–i): low-frequency filter (LFF), 1 Hz; high-frequency filter (HFF), 70 Hz; amplitude, 150 P–P.
Patient was initially started on levetiracetam, at a rapid dose escalation, and topiramate, which were not effective. High-dose adrenocorticotropic hormone (ACTH) was initiated and infantile spasms rapidly subsided. The EEG recorded 2 weeks later showed resolution of the hypsarrhythmia. The only remaining EEG abnormalities were intermittent, multifocal spikes with posterior dominance (Figures 1f and g). An EEG 3 months later revealed deterioration of the background activity with an increase in the frequency of high amplitude spikes, creating hypsarrhythmic pattern during sleep. No clinical seizures were observed. Vigabatrin was started and the EEG’s background activity slowly improved. Vigabatrin was discontinued around 28 months of age and he remains seizure free. EEG at 33 months of age (Figures 1h and i) showed a slow, disorganized background with frequent multifocal spikes demonstrating occipital dominance; however, hypsarrhythmia was not present.
As of 33 months of age, the child has progressed with development but has delayed milestones. He was able to sit unsupported by 18 months, crawl at 27 months, and began walking independently at 31 months of age. Communication is limited to his ability to utter vowel sounds.
The proband’s younger brother presented at 1 month of age with remarkable clinical differences to the proband. He had normal intrauterine movements and was not hypotonic at the time of the evaluation. Despite his normal exam, EEG at 1 month showed frequent occipital polyspikes, in addition to less frequent multifocal discharges. At 2 months of age, he developed episodes of subtle head movements with associated upward eye deviation. These events occurred in clusters and were associated with ‘electrodecrements’ in the EEG, consistent with ‘subtle spasms’. His EEG did not show hypsarrhythmia. Early treatment with vigabatrin completely controlled the clinical events. Last follow-up at 5 months of age revealed visual inattention and lack of face tracking despite normal tone. An MRI had not been obtained at the time of publication.
Molecular analysis of the ARX variant
Genomic DNA from the proband was extracted from the peripheral blood lymphocytes using the AutoGen-automated DNA isolation system (Autogen Inc., Holliston, MA, USA), following the manufacturer’s recommendations. All five coding exons3 and flanking intron sequences of the ARX gene were amplified by polymerase chain reaction (PCR) and sequenced bidirectionally on an ABI 3730xl sequencer (Life-Technologies, Carlsbad, CA, USA). For the brother of the proband, PCR amplification and bidirectional sequencing of ARX exon 1 was performed on genomic DNA extracted from peripheral blood lymphocytes. Sequence analysis was performed using Mutation Surveyor (Soft Genetics Inc., State College, PA, USA). The ARX cDNA reference sequence used was NM_139058.2.
Cloning of mutant full-length ARX constructs
Full-length human ARX cDNA construct in pCMV-Myc vector (pCMV-Myc-ARX Wt) was obtained.29 To generate the mutant ARX constructs, site-directed mutagenesis (QuikChange Multi Site-Directed Mutagenesis Kit; Agilent Technologies, Santa Clara, CA, USA) was used to introduce single-nucleotide variation into pCMV-Myc-ARX Wt following the manufacturer’s instructions. The following mutant constructs were generated: pCMV-Myc-ARX c.34G>T (p.E12*), pCMV-Myc-ARX c.122T>G (p.M41R), pCMV-Myc-ARX c.181G>T (p.E61*), and pCMV-Myc-ARX c.(34G>T; 122T>G) (p.E12*-M41R). The primer sequences are available upon request. The mutant construct pCMV-Myc-ARX Y27* was a gift from Prof. Jozef Gecz (University of Adelaide, Adelaide, SA, Australia). The entire open reading frames (ORFs) of all constructs were verified by sequencing to ensure no other variation was introduced. All cDNA clones and subsequently expressed protein will be described by the protein variation nomenclature, that is, pCMV-Myc-ARX c.34G>T(p.E12*) will be referred to as p.(E12*).
Cell culture, transient transfection, and immunofluorescence
Cell culture and transfection of HEK293T cells with plasmid DNA were as described in Shoubridge et al.30 Transfection rates were determined by the amount of ARX-positive cells in an average of 300 cells. ARX-positive cells were detected via immunofluorescence as described previously.30
Protein extraction, SDS-PAGE, and immunoblot
Transfected cells were harvested 24 h posttransfection and lysed in radioimmunoprecipitation assay buffer (65.3 mm Tris (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 1 mm NaVO3, 1 mm NaF and 1x protease inhibitor cocktail; Sigma-Aldrich, Castle Hill, NSW, Australia). Insoluble material was removed by centrifugation at 11 000 g for 15 min. To determine the equivalent amounts of protein expression for wild-type and mutant ARX, cleared lysates were separated on a 4–12% Bis-Tris gel (Life-Technologies, Melbourne, VIC, Australia) and transferred onto BioTrace NT nitrocellulose membranes (Pall Life Sciences, Cheltenham, VIC, Australia). Membranes were blocked with 5% skim milk, incubated with either mouse anti-ARX (1 μg/ml final),29 mouse anti-cMyc (1:8000) (9E10; Santa Cruz Biotechnology, Dallas, TX, USA), or mouse anti-β-actin (1:50 000) (AC74; Sigma-Aldrich), and subsequently with goat anti-mouse IgG HRP-conjugated (1:2000) (Dako Australia, Sydney, NSW, Australia). Signal was detected by chemiluminescence.
Luciferase reporter assays
Protocols were performed as described previously.31 In brief, we generated a luciferase reporter construct of orthologous sequence upstream of human LMO1 gene containing specific transcription factor binding sites for ARX directionally cloned upstream of the SV40 promoter driving the luciferase reporter gene (luc2) in the pGL4.13[luc2/SV40] vector (Promega, Madison, WI, USA). Cells were harvested 24 h posttransfection with 500 ng luciferase reporter plasmid DNA, 10 ng pGL4.74[hRluc/TK] plasmid DNA (Promega) and variable amounts of Myc expression plasmid DNA containing full-length ARX-Wt or premature truncation mutation sequence using Lipofectamine 2000 (Life-Technologies). Firefly and Renilla luciferase was achieved using Dual-Glo Luciferase Assay System (Promega) on a Veritus Microplate Luminometer (Turner BioSystems, Sunnyvale, CA, USA). In three independent transfections, each sample was carried out in replicate, with triplicates of each replicate measured in the reporter assay. The firefly luciferase activity was normalized according to the corresponding Renilla luciferase activity, and the ratio of luciferase activity was reported relative to pCMV-Myc empty vector for each transfection. Wilcoxon–Mann–Whitney U-test was performed to examine differences between the comparisons of equivalent plasmid and protein.
Results
Molecular analysis of the ARX gene
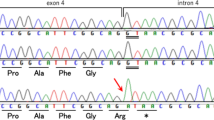
The diagnoses of infantile spasms or West syndrome in the proband prompted the evaluation of the ARX gene. Sequence analysis demonstrated a novel nucleotide change, c.34G>T in exon 1 of the ARX gene, that resulted in the creation of a premature termination codon, p.(E12*) (Figure 2). The child’s mother was confirmed to be a heterozygous carrier for the same variant. The proband’s younger brother was also determined to be hemizygous for the c.34G>T (p.(E12*)) change.

Pedigree and molecular analysis. The proband is indicated with a bold black arrow. The c.34G>T (p.(E12*)) pathogenic sequence change seen in the hemizygous state in both affected brothers and in the carrier state in their mother is depicted under each individual with an arrow. A normal sequence is also included for comparison.
ARX mRNA containing the premature protein termination variation c.34G>T (p.(E12*)) produces an N-terminal- truncated ARX protein
The c.34G>T nucleotide change is predicted to substitute the glutamic acid (p.E12) encoded by GAG to UAG, that codes for a termination codon, which results in the NMD of the mutant transcript, and no functional ARX protein. Given the similar phenotype of our patient to the individuals with the p.(Y27*) variation,2 we predicted that the premature protein termination variation (c.34G>T (p.(E12*))) would reinitiate mRNA translation, potentially at p.M41, and produce a truncated protein missing the N-terminal region (Figure 3a). Following overexpression of the mutant forms of human ARX cDNA constructs, we observed a product migrating below the full-length ARX protein, but only when detected by an ARX antibody29 and not when using an antibody against the N-terminal fused Myc-tag (Figure 3b), indicating that an N-terminally truncated protein was produced. The truncated protein migrated at the same size as that of the truncated protein from p.(Y27*), suggesting that reinitiation at p.M41 occurs for both mutant proteins.

Reinitiation of mRNA translation after premature N-terminal protein termination mutations in ARX and potential reinitiation sites in the N-terminal region of ARX. (a) Schematic of the human ARX protein. Human ARX domains and regions are indicated above the schematic and their locations are indicated below. Known functional domains are highlighted, octapeptide (OP) as horizontally hatched rectangle, nuclear localization sequences (NLS) as three black rectangles, polyalanine tracts (PA) as four white rectangles, acidic domain as vertically hatched rectangle, homeodomain crosshatched, and aristaless domain (OAR) hatched. Myc tag is displayed as a smaller gray square. For Wt and mutant ARX (listed on the left), the translation of each Myc-tagged ARX is indicated by the black arrows with predicted termination of translation at the black ‘stop’ signs for each. Predicted reinitiation of translation is indicated by gray arrows for the (Y27*) and (E12*) mutations. The regions detected by the Myc and ARX antibodies are shown as small black rectangles below the protein, with gray bars indicating each epitope location on each of the predicted proteins. The amino-acid length of each product is indicated below each protein. The star indicates the predicted residue (p.M41) at which reinitiation of translation is likely to occur. The table on the right details the predicted size of the resulting protein produced from overexpression from a CMV promoter in transiently transfected HEK293T cells as detected by antibodies against the Myc (left) or ARX (right) epitopes. (b) Following 24 h of overexpression in HEK293T cells, protein lysates were subjected to SDS-PAGE and immunoblotted with antibodies against amino acids 161–179 of ARX ORF (top panel) or against the Myc tag (bottom panel). The ARX-Wt (lane 1) and p.M41R (lane 4) samples were loaded with 2 μg of total protein lysate. The p.(E12*) (lane 2) was loaded with 10 μg of protein lysate, with p.(Y27*) (lane 3), p.E61X (lane 5), p.E12*-M41R (lane 6), and non-transfected (lane 7) loaded with 20 μg of protein lysate. β-Actin was probed on each membrane and highlights the different amounts of protein loaded in each lane. The result is representative of three independent experiments. (c) The first 630 nucleotides in ARX are shown as individual codons and the corresponding amino acids (1–210) listed above each codon. The translation start site p.M1 is highlighted in red, as well as reinitiation site p.M41, and potential sites at p.L54, p.V64,p.L79, p.L81, p.L85, and p.L88. The region used to generate the ARX antibody is shown in the green box. p.E61 is shown by a crossed rectangle. Alternative start codons besides the most common start codon AUG (M) are stated at the bottom of the figure.
To test if reinitiation was occurring at this residue, we engineered the p.M41 to an arginine residue (p.M41R) alone and in the presence of p.(E12*). The alteration of this residue to an arginine alone, and in the absence of the truncating variant, resulted in the production of a full-length protein. Although we predicted that removal of methionine at p.41 would abolish protein reinitiation when present together with the p.(E12*) variant, a protein smaller than either the p.(E12*) or the p.(Y27*) variants alone was observed (Figure 3b, lane 6). As this smaller, N-terminally truncated protein was detected with the ARX antibody, which detects an epitope between amino acids 161 and 179 of ARX, we reasoned that the more distant reinitiation must have occurred before this region. Our analysis of the amino-acid sequence identified that there are no other codons for methionine between p.M41 and the epitope recognition site ending at p.G179 (Figure 3c). However, other non-AUG codons, for example, those for lysine can be used as alternative start codons. There were several of these non-AUG alternative start codons within this region. Given the size of the migrating band in the E12*-M41R mutant sample, we predicted that the second initiation site may be via a non-Aug codon at p.L54, encoded by a CTG. We were interested to see at what point a premature termination variant would no longer lead to reinitiation. Engineering a stop codon at p.E61 resulted in no detectable protein using the anti-ARX antibody (Figure 3b), but did generate a 10 kDa band detectable with the anti-Myc antibody corresponding to the truncated protein p.M1_P60 (data not shown).
Early truncation variants in ARX markedly reduced the levels of expressed protein and in turn reduced the transcriptional repression activity
Although we showed in an in vitro setting that reinitiation of protein translation does occur following early termination variants p.(E12*) and p.(Y27*) in ARX, the levels of the truncated protein were markedly reduced, requiring higher amounts of protein lysate to be loaded per lane to detect the protein. Compared with wild-type ARX, up to 5x p.(E12*) and 10x p.(Y27*) more protein lysates were loaded (Figure 3b). The differences between ARX-Wt and p.(E12*) were apparent even in the presence of similar rates of transient transfection. Compared with ARX-Wt and p.(E12*), there were consistently lower number of cells expressing truncated ARX protein following transfection with the p.(Y27*) plasmid, detected by either immunofluorescence (data not shown) or western immunoblotting. It remains unclear to what extent this reduction in protein expression is due to compromised reinitiation efficiency. We examined an equivalent amount of protein lysates across all samples taking into account the variations in transfection efficiency. For example, we identified that 2 μg of ARX-Wt from a population of cells with a 30% transfection rate would give a robust signal by immunoblot. As the transfection rate for p.(E12*) was 37%, we loaded 1.6 μg (Figure 4a). We also loaded 6 μg of ARX-Wt and the equivalent of each mutant lysate to capture the less abundant proteins (Figure 4a). Using this approach, we determined that p.(E12*) was markedly reduced in expression compared with wild type, whereas p.(Y27*) and the double variant p.E12*-M41R could not be detected (Figure 4a).

Impact of protein expression compared with the loss of N terminus on mutant protein function. (a) HEK293T cells were transfected with a set amount (0.5 μg) of plasmid DNA, and 24 h posttransfection the resulting protein lysates were subjected to SDS-PAGE and immunoblotted for ARX protein. In all, 6 and 2 μg of protein lysate was loaded for ARX-Wt. Taking into account the variations in transfection rate (listed below the figure), equivalent amounts of protein lysate were loaded for all other samples. (b) HEK293T cells transfected with diminishing concentrations of plasmid DNA and a set amount (2 μg) of protein lysate was loaded per lane. Results from two independent experiments are shown. The stars indicate equivalent signal strength of bands of the expressed proteins. β-Actin was probed on each membrane to detect the amounts of protein loaded in each lane. (c) The luciferase reporter assay used three copies of the ARX-binding site identified in the enhancer region of Lmo1, cloned upstream of SV40 luciferase. Luciferase data were normalized to Renilla expression and expressed as a percentage value relative to expression in empty Myc-vector-transfected cells. Myc-tagged constructs and amounts transfected (in μg) are listed along the bottom of the graph: Myc vector (white), ARX-Wt (black to gray), and ARX (E12*) (red to light red). The bars at the bottom of graph indicate the amount of plasmid DNA transfected. The dotted line marks the similar levels of transcriptional repression of luciferase expression achieved when 0.25 μg of ARX-Wt plasmid and 1.0 μg of (E12*) plasmid are transfected. Error bars indicate s.e.m. from three independent experiments. (d) Data from the luciferase assays are shown with equivalent plasmid transfected compared with equivalent amounts of expressed protein. *P<0.05.
Early truncation variants in ARX substantially impact the function of the protein as evidenced by the severe clinical phenotype of the affected patients. However, what is less clear is the relative impact of the loss of the N terminus compared with the predicted reduction in protein expression. To address this issue, we transfected diminishing concentrations of plasmid DNA into HEK293T cells and determined the relative levels of protein expression by immunoblot and compared this with the corresponding transcriptional repression activity using a luciferase reporter assay. ARX-Wt routinely represses luciferase expression when the regulatory sequence of human Lmo1, a known gene repression target of ARX, was inserted upstream of firefly luciferase reporter. Our analysis showed a band of similar signal strength for p.(E12*) transfected at 0.5 μg to that seen with ARX-Wt from between 0.25 and 0.125 μg transfected across replicate experiments (Figure 4b). This indicates that approximately half to a third as much mutant p.(E12*) protein was expressed compared with full-length wild-type for the same amount of plasmid DNA transfected. Transfection of diminishing amounts of plasmid DNA of ARX-Wt resulted in a dose-dependent decrease in the repression activity in the luciferase reporter assay (Figure 4c). A partial loss of repression activity was observed from ARX p.(E12*) (Figures 4c), p.(Y27*), and p.E12*-M41R (data not shown). Using these data we can capture the functional differences because of the lack of the N terminus compared with the relative reduction of protein expressed. When we compare the same amount of plasmid DNA transfected, we see a significant 1.3-fold loss of transcriptional repression with p.(E12*) as compared with ARX-Wt (Figure 4d). However, when we corrected for equivalent amounts of expressed protein, we noted that the capacity of the p.(E12*) mutant to transcriptionally repress expression in our reporter assay was similar to ARX-Wt. Taken together, these data suggest that the capacity of the N-terminally truncated protein is impacted not only by the loss of the octapeptide domain but is also impaired because of the reduction in protein expression, likely due to the inefficient process of reinitiation.
Discussion
Here we present two brothers with a novel premature protein termination (PTC) variation (c.34G>T/ p.(E12*)) in ARX. Such a PTC variant would be predicted to give rise to a catastrophic XLAG phenotype because of the loss of ARX function. Despite this, we report the relatively milder clinical phenotype of ISSX, giving rise to an inconsistent genotype–phenotype correlation. Upon in vitro transfection studies, we detected the presence of N-terminally truncated ARX protein in cell lysates overexpressing the ARX p.(E12*) construct, suggesting reinitiation of translation after the premature termination codon. This is the second family with early premature protein termination variation in ARX that results in reinitiation of protein translation producing a partially functional N-terminally truncated ARX protein.2 Both cases reported milder phenotypes compared with the expected XLAG, indicating that the N-terminally truncated protein is able to partially function and ‘rescue’ the patients from the brain malformation phenotype.
Reinitiation of translation refers to the mechanism that allows the ribosome to bind and initiate at a downstream start codon after a stop codon has been met upstream32, 33, 34, 35 and reviewed in Jackson et al.36 A substantial number (>45%) of mammalian genes contain at least one short upstream ORF (upORF) preceding the coding domain of the gene.34, 36 The process of reinitiation is often inefficient. The efficiency decreases when ribosome elongation time is lengthened during the translation of upORF, either due to the length of upORF or the presence of pseudoknot in the upORF.34 Efficiency of reinitiation is also improved by increasing the distance between the termination codon and downstream start codon, possibly due to the rebinding of Met-tRNA.33 Hence, the region before the PTC in our patients would act as an upORF, consistent with our observation of markedly reduced abundance compared with ARX-Wt with p.(E12*) and even lower abundance with p.(Y27*). It is unclear to what extent this reduction in protein expression occurs in the patients, as ARX is not expressed adequately in available patient material. We cannot exclude that the reduction in protein abundance of the N-terminally truncated proteins may also be due in part to increased susceptibility to NMD or altered stability of the truncated protein itself.
NMD is a posttranscriptional surveillance mechanism that detects and degrades aberrant mRNA transcript containing PTC to prevent the translation of aberrant protein.37 Cases of premature protein termination variation early in the gene followed by reinitiation of translation at a downstream start codon in other genes are not rare, suggesting an alternative to escape NMD.38,39 In many cases of reinitiation, N-terminally truncated proteins are predicted to rescue the patient from a more severe loss-of-function phenotype, including truncating variants within the first exon of DMD gene, resulting in the milder Becker muscular dystrophy instead of the predicted more severe Duchenne muscular dystrophy (DMD).40
In ARX, both the N-terminal octapeptide domain and the region flanking polyalanine tract 4 function as repressor domains.41 The octapeptide domain shows repression activity using in vitro reporter assays, but at a reduced extent compared with the full-length protein.41 This highly conserved domain shares homology with the Engrailed homology 1 domain, a repressor domain in the Drosophila homeobox protein Engrailed.42 The octapeptide domain in ARX binds directly to Groucho’s mammalian homolog Transducin-like enhancer of split 1 (TLE1) protein.41 The presence of TLE1, 2 or 4 proteins enhances the repression activity of ARX.41 In brain development, Gro/TLE proteins expressed in proliferating neural progenitor cells have been found to be involved in neurogenesis through the maintenance of the undifferentiated state by inhibiting or delaying neuronal differentiation.43, 44 A missense mutation in the conserved leucine residue to proline (c.98T>C/p.(L33P)) in the octapeptide domain of ARX has been identified in a family affected with non-syndromic ID.45 The transcription repression activity of ARX containing the (c.98T>C/p.(L33P)) mutation was compromised because of an inability of the mutant to bind to TLE1 protein.41 Hence, a loss of the octapeptide domain because of N-terminal truncation would compromise the repression activities regulated by this domain.
Our analysis indicates that a reduction in protein abundance is also likely to contribute to the altered transcriptional activity of the N-terminally truncated mutant protein. We have growing evidence from other mutations in ARX that a reduction in protein abundance may be a key contributor to pathogenesis. We reported a significant reduction of ARX protein abundance in mice modeling the two most common polyalanine tract expansion mutations.46 Adequate repression of Lmo1, a direct target gene of ARX47, 48, 49 was compromised in the brains of these mice during early development.46 A significant loss of LMO1 repression earlier in development was noted with the more severe polyalanine tract 1 mutation, and correlates with clinical severity displayed by patients with these polyalanine tract mutations. Interestingly, both the c.34G>T (p.(E12*) patient in this current study and the c.81C>G (p.(Y27*) patients display similar clinical features to patients with polyalanine tract expansion mutations. Given the restricted expression pattern of ARX, we were unable to test the levels of ARX protein abundance in our patients.
Conclusions
We have identified a novel c.34G>T (p.(E12*) variant in ARX in a pair of brothers presenting with early-onset infantile spasms instead of the more severe malformation phenotype of XLAG. We demonstrate that this premature termination variant actually leads to reinitiation of protein translation producing an N-terminally truncated ARX protein at a reduced abundance. We propose that the loss of the octapeptide domain in conjunction with low protein abundance negatively impacts ARX transcriptional activity contributing to the disease pathogenesis in our patients.
References
Shoubridge C, Fullston T, Gecz J : ARX spectrum disorders: making inroads into the molecular pathology. Hum Mutat 2010; 31: 889–900.
Fullston T, Brueton L, Willis T et al: Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X). Eur J Hum Genet 2010; 18: 157–162.
Stromme P, Mangelsdorf ME, Shaw MA et al: Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet 2002; 30: 441–445.
Kato M, Das S, Petras K, Sawaishi Y, Dobyns WB : Polyalanine expansion of ARX associated with cryptogenic West syndrome. Neurology 2003; 61: 267–276.
Wohlrab G, Uyanik G, Gross C et al: Familial West syndrome and dystonia caused by an Aristaless related homeobox gene mutation. Eur J Pediatr 2005; 164: 326–328.
Guerrini R, Moro F, Kato M et al: Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 2007; 69: 427–433.
Poirier K, Eisermann M, Caubel I et al: Combination of infantile spasms, non-epileptic seizures and complex movement disorder: a new case of ARX-related epilepsy. Epilepsy Res 2008; 80: 224–228.
Wallerstein R, Sugalski R, Cohn L, Jawetz R, Friez M : Expansion of the ARX spectrum. Clin Neurol Neurosurg 2008; 110: 631–634.
Reish O, Fullston T, Regev M, Heyman E, Gecz J : A novel de novo 27bp duplication of the ARX gene, resulting from postzygotic mosaicism and leading to three severely affected males in two generations. Am J Med Genet Part A 2009; 149A: 1655–1660.
Cossee M, Faivre L, Philippe C et al: ARX polyalanine expansions are highly implicated in familial cases of mental retardation with infantile epilepsy and/or hand dystonia. Am J Med Genet Part A 2011; 155A: 98–105.
Conti V, Marini C, Gana S, Sudi J, Dobyns WB, Guerrini R : Corpus callosum agenesis, severe mental retardation, epilepsy, and dyskinetic quadriparesis due to a novel mutation in the homeodomain of ARX. Am J Med Genet Part A 2011; 155A: 892–897.
Fullston T, Finnis M, Hackett A et al: Screening and cell-based assessment of mutations in the Aristaless-related homeobox (ARX) gene. Clin Genet 2011; 80: 510–522.
Mirzaa GM, Paciorkowski AR, Marsh ED et al: CDKL5 and ARX mutations in males with early-onset epilepsy. Pediatr Neurol 2013; 48: 367–377.
Kato M, Saitoh S, Kamei A et al: A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet 2007; 81: 361–366.
Absoud M, Parr JR, Halliday D, Pretorius P, Zaiwalla Z, Jayawant S : A novel ARX phenotype: rapid neurodegeneration with Ohtahara syndrome and a dyskinetic movement disorder. Dev Med Child Neurol 2010; 52: 305–307.
Eksioglu YZ, Pong AW, Takeoka M : A novel mutation in the aristaless domain of the ARX gene leads to Ohtahara syndrome, global developmental delay, and ambiguous genitalia in males and neuropsychiatric disorders in females. Epilepsia 2011; 52: 984–992.
Kato M, Koyama N, Ohta M, Miura K, Hayasaka K : Frameshift mutations of the ARX gene in familial Ohtahara syndrome. Epilepsia 2010; 51: 1679–1684.
Giordano L, Sartori S, Russo S et al: Familial Ohtahara syndrome due to a novel ARX gene mutation. Am J Med Genet Part A 2010; 152A: 3133–3137.
Sartori S, Polli R, Bettella E et al: Pathogenic role of the X-linked cyclin-dependent kinase-like 5 and aristaless-related homeobox genes in epileptic encephalopathy of unknown etiology with onset in the first year of life. J Child Neurol 2011; 26: 683–691.
Kato M, Das S, Petras K et al: Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Human mutation 2004; 23: 147–159.
Kitamura K, Yanazawa M, Sugiyama N et al: Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nature genetics 2002; 32: 359–369.
Uyanik G, Aigner L, Martin P et al: ARX mutations in X-linked lissencephaly with abnormal genitalia. Neurology 2003; 61: 232–235.
Hahn A, Gross C, Uyanik G et al: X-linked lissencephaly with abnormal genitalia associated with renal phosphate wasting. Neuropediatrics 2004; 35: 202–205.
Hartmann H, Uyanik G, Gross C et al: Agenesis of the corpus callosum, abnormal genitalia and intractable epilepsy due to a novel familial mutation in the Aristaless-related homeobox gene. Neuropediatrics 2004; 35: 157–160.
Bhat SS, Rogers RC, Holden KR, Srivastava AK : A novel in-frame deletion in ARX is associated with lissencephaly with absent corpus callosum and hypoplastic genitalia. Am J Med Genet Part A 2005; 138: 70–72.
Okazaki S, Ohsawa M, Kuki I et al: Aristaless-related homeobox gene disruption leads to abnormal distribution of GABAergic interneurons in human neocortex: evidence based on a case of X-linked lissencephaly with abnormal genitalia (XLAG). Acta Neuropathol 2008; 116: 453–462.
Miyata R, Hayashi M, Miyai K, Akashi T, Kato M, Kohyama J : Analysis of the hypothalamus in a case of X-linked lissencephaly with abnormal genitalia (XLAG). Brain Dev 2009; 31: 456–460.
Oegema R, Maat-Kievit A, Lequin MH et al: Asymmetric polymicrogyria and periventricular nodular heterotopia due to mutation in ARX. Am J Med Genet Part A 2012; 158A: 1472–1476.
Shoubridge C, Cloosterman D, Parkinson-Lawerence E, Brooks D, Gecz J : Molecular pathology of expanded polyalanine tract mutations in the Aristaless-related homeobox gene. Genomics 2007; 90: 59–71.
Shoubridge C, Tan MH, Fullston T et al: Mutations in the nuclear localization sequence of the Aristaless related homeobox; sequestration of mutant ARX with IPO13 disrupts normal subcellular distribution of the transcription factor and retards cell division. PathoGenetics 2010; 3: 1.
Shoubridge C, Tan MH, Seiboth G, Gecz J : ARX homeodomain mutations abolish DNA binding and lead to a loss of transcriptional repression. Hum Mol Genet 2012; 21: 1639–1647.
Kozak M : Selection of initiation sites by eucaryotic ribosomes: effect of inserting AUG triplets upstream from the coding sequence for preproinsulin. Nucleic Acids Res 1984; 12: 3873–3893.
Kozak M : Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol Cell Biol 1987; 7: 3438–3445.
Kozak M : Constraints on reinitiation of translation in mammals. Nucleic Acids Res 2001; 29: 5226–5232.
Skabkin MA, Skabkina OV, Hellen CU, Pestova TV : Reinitiation and other unconventional posttermination events during eukaryotic translation. Mol Cell 2013; 51: 249–264.
Jackson RJ, Hellen CU, Pestova TV : Termination and post-termination events in eukaryotic translation. Adv Protein Chem Struct Biol 2012; 86: 45–93.
Popp MW, Maquat LE : Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet 2013; 47: 139–165.
Howard MT, Malik N, Anderson CB, Voskuil JL, Atkins JF, Gibbons RJ : Attenuation of an amino-terminal premature stop codon mutation in the ATRX gene by an alternative mode of translational initiation. J Med Genet 2004; 41: 951–956.
Buisson M, Anczukow O, Zetoune AB, Ware MD, Mazoyer S : The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum Mut 2006; 27: 1024–1029.
Gurvich OL, Maiti B, Weiss RB, Aggarwal G, Howard MT, Flanigan KM : DMD exon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Hum Mut 2009; 30: 633–640.
McKenzie O, Ponte I, Mangelsdorf M et al: Aristaless-related homeobox gene, the gene responsible for West syndrome and related disorders, is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. Neuroscience 2007; 146: 236–247.
Fisher AL, Caudy M : Groucho proteins: transcriptional corepressors for specific subsets of DNA-binding transcription factors in vertebrates and invertebrates. Genes Dev 1998; 12: 1931–1940.
Buscarlet M, Hermann R, Lo R, Tang Y, Joachim K, Stifani S : Cofactor-activated phosphorylation is required for inhibition of cortical neuron differentiation by Groucho/TLE1. PLoS One 2009; 4: e8107.
Yao J, Liu Y, Lo R, Tretjakoff I, Peterson A, Stifani S : Disrupted development of the cerebral hemispheres in transgenic mice expressing the mammalian Groucho homologue transducin-like-enhancer of split 1 in postmitotic neurons. Mech Dev 2000; 93: 105–115.
Bienvenu T, Poirier K, Friocourt G et al: ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002; 11: 981–991.
Lee K, Mattiske T, Kitamura K, Gecz J, Shoubridge C : Reduced polyalanine-expanded Arx mutant protein in developing mouse subpallium alters Lmo1 transcriptional regulation. Hum Mol Genet 2014; 23: 1084–1094.
Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA : Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet 2008; 17: 3740–3760.
Friocourt G, Parnavelas JG : Identification of Arx targets unveils new candidates for controlling cortical interneuron migration and differentiation. Front Cell Neurosci 2011; 5: 28.
Colasante G, Sessa A, Crispi S et al: Arx acts as a regional key selector gene in the ventral telencephalon mainly through its transcriptional repression activity. Dev Biol 2009; 334: 59–71.
Acknowledgements
We thank the patients, their families and physicians for their participation in this study. We also thank Nancy Briggs from the Robinson Research Institute, University of Adelaide for the statistical analysis. The Neurogenetics research program in the Department of Paediatrics, University of Adelaide, Australia was funded by the Australian National Health and Medical Research Council (Grant No. 1063025). CS is supported Australian Research Council (Future Fellowship FT120100086).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Moey, C., Topper, S., Karn, M. et al. Reinitiation of mRNA translation in a patient with X-linked infantile spasms with a protein-truncating variant in ARX. Eur J Hum Genet 24, 681–689 (2016). https://doi.org/10.1038/ejhg.2015.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.176
