
Abstract
Coenzyme Q10 (CoQ10) has an important role in mitochondrial energy metabolism by way of its functioning as an electron carrier in the respiratory chain. Genetic defects disrupting the endogenous biosynthesis pathway of CoQ10 may lead to severe metabolic disorders with onset in early childhood. Using exome sequencing in a child with fatal neonatal lactic acidosis and encephalopathy, we identified a homozygous loss-of-function variant in COQ9. Functional studies in patient fibroblasts showed that the absence of the COQ9 protein was concomitant with a strong reduction of COQ7, leading to a significant accumulation of the substrate of COQ7, 6-demethoxy ubiquinone10. At the same time, the total amount of CoQ10 was severely reduced, which was reflected in a significant decrease of mitochondrial respiratory chain succinate-cytochrome c oxidoreductase (complex II/III) activity. Lentiviral expression of COQ9 restored all these parameters, confirming the causal role of the variant. Our report on the second COQ9 patient expands the clinical spectrum associated with COQ9 variants, indicating the importance of COQ9 already during prenatal development. Moreover, the rescue of cellular CoQ10 levels and respiratory chain complex activities by CoQ10 supplementation points to the importance of an early diagnosis and immediate treatment.
Similar content being viewed by others

Introduction
Coenzyme Q10 (CoQ10) is a lipid-soluble, naturally occurring substance, which functions as an essential cofactor of the mitochondrial respiratory chain.1 It acts as an electron carrier from complex I and complex II to complex III and thereby goes through a redox cycle between the oxidized (ubiquinone) and the reduced form (ubiquinol). Moreover, CoQ10 links mitochondrial fatty acid β-oxidation to the respiratory chain, participates in the biosynthesis of pyrimidines and modulates apoptosis.2 In its reduced form the CoQ10 molecule serves as a potent antioxidant.
CoQ10 is acquired through biosynthesis or dietary intake. However, biosynthesis is the major source of CoQ10, which involves the coordinated function of at least 13 genes (PDSS1, PDSS2, COQ2, COQ3, COQ4, COQ5, COQ6, COQ7, ADCK3, ADCK4, COQ9, COQ10A and COQ10B). To date, defects in eight of these genes were identified as a cause of primary CoQ10 deficiency disorders (PDSS1, PDSS2, COQ2, COQ4, COQ6, ADCK3, ADCK4 and COQ9), a clinically heterogeneous group of diseases, which frequently manifests in childhood.3, 4 According to Quinzii et al.,5 five major clinical phenotypes can be distinguished: (1) encephalomyopathy, (2) severe infantile multisystemic disease, (3) nephropathy, (4) cerebellar ataxia and (5) isolated myopathy. Identification of CoQ10 deficiency is important because CoQ10 supplementation can be beneficial in certain conditions.
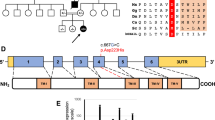
Until now, just a single patient with CoQ10 deficiency caused by a homozygous stop variant in COQ9 c.730C>T, p.Arg244*, leading to neonatal lactic acidosis, intractable seizures and global developmental delay, has been reported.6 Using exome sequencing, we identified a new variant affecting function in COQ9 associated with neonatal encephalopathy and early death. Our report expands the clinical spectrum associated with COQ9 variants and indicates a severe pre-/neonatal-onset phenotype. COQ9-deficient patient fibroblasts were characterized in detail and the functional consequences of the genetic defect are shown.
Case report
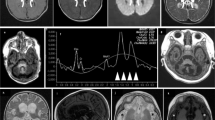
The boy reported here was the second child of healthy, first-grade consanguineous Turkish parents. His 13-year-old brother is healthy. Pregnancy was normal until the 27th gestational week when oligohydramnion was noticed. At that time the parents declined a recommended cesarean section. Finally, the child was born at 36 weeks of gestation by cesarean section due to anhydramnion. APGAR scores were 5/4/6, respectively. The neonate was small for gestational age with a birth weight of 1440 g, length of 39.5 cm and head circumference of 28.8 cm (all values below 3rd percentile). No dysmorphic stigmata were observed. During the immediate postnatal period the patient showed poor respiratory efforts, muscular hypotonia, bradycardia and generalized cyanosis. The child was intubated and transferred to the neonatal intensive care unit. Laboratory investigations revealed lactic acidosis (up to 22 mmol/l; norm: <1.8 mmol/l), requiring continuous buffering therapy with sodium bicarbonate. Cranial ultrasound demonstrated multiple choroid plexus cysts and symmetrical hyperechoic signal alterations in the basal ganglia, suggesting neonatal Leigh-like syndrome (Figure 1a). During the following 4 days, lactate levels remained elevated (up to 26 mmol/l) despite intravenous glucose infusion and buffering therapy. Laboratory investigations revealed elevated blood alanine (1088 μmol/l, norm <414 μmol/l) and massive lactic-acid excretion in urine. On the fifth day of life, slow improvement in acidosis was noticed with minimal lactate levels of 5 mmol/l. Respiratory efforts improved and the boy was extubated with adequate spontaneous breathing on the eighth day of life. However, the child showed reduced spontaneous movements with intermittent opisthotonus and muscle stiffness in the extremities. On the tenth day of life, the child suffered from seizures and recurrent episodes of apnea and bradycardia. The boy’s clinical condition further deteriorated and he died due to cardio-respiratory failure at 18 days of age. Because of a suspected mitochondrial disease, a skin biopsy was performed. Activity measurements of the oxidative phosphorylation enzymes in cultured fibroblasts showed a strongly reduced activity of respiratory chain succinate-cytochrome c oxidoreductase (complex II+III: 62 mU/UCOX, norm>160), pointing to a CoQ10 metabolism defect. Histological investigations of muscle and liver tissue were without specific findings.

(a) Brain ultrasound in a newborn with a pathogenic COQ9 variant. Left and middle panels: right and left parasagittal view, respectively, showing hyperechoic signal within the basal ganglia (arrow) and bilateral choroid plexus cysts (asterisk). Right panel: coronal view demonstrating symmetrical hyperechoic basal ganglia abnormalities suggesting a neonatal Leigh-like syndrome. (b) Schematic diagram showing the transcript generated in the presence of the c.521+1 deletion. Chromatogram shows that the deletion causes a skipping of exons 4 and 5 in the patient fibroblasts. (c) Western blotting on mitochondria-enriched fractions of patient and control fibroblasts (representative image). In patient cells no COQ9 is detected in the non-transduced condition (−). Signal is restored after lentiviral transduction with COQ9 wild-type cDNA (+). In addition, a COQ7 antibody was used, demonstrating a severe reduction of COQ7 protein in patient cells. Porin (VDAC) was used as a loading marker.
Materials and methods
A detailed description of the materials and methods used for this paper can be found in the Supplementary Material.
Results and discussion
To unravel the genetic cause of the disease we performed exome sequencing as described previously.7 Assuming an autosomal-recessive mode of inheritance, our analyses focused on homozygous and predictively compound heterozygous nonsynonymous variants with a MAF<0.1% in 4500 control exomes. This search identified 28 genes. Only one of these genes, COQ9 (NM_020312.2; NG_027696.1), coded for a mitochondrial protein.8 In addition, COQ9 was the only gene carrying two predicted loss-of-function alleles. The identified splice site variant, c.521+1del, p.(Ser127_Arg202del), was confirmed by Sanger sequencing in the homozygous state in the index patient with the parents being heterozygous carriers. The change c.521+1del is absent from 4500 in-house control exomes and is listed only once in the heterozygous state in 111 520 alleles of the Exome Aggregation Consortium Browser (Cambridge, MA, USA;http://exac.broadinstitute.org; 12/2014). The c.521+1 deletion is likely to significantly reduce the efficiency of the donor splice site of intron 4, with the most likely effect being skipping of exon 4 during splicing. Indeed, sequencing of the COQ9 cDNA showed a loss of exons 4 and 5 in patient fibroblasts (Figure 1b). The predicted consequence of the variant on the COQ9 protein is a deletion of 76 amino acids, including essential parts of the lipid binding cavity.9
We next investigated the effect of the variant on the expression of COQ9 by immunodecoration of mitochondria-enriched proteins using a COQ9-specific antibody. Using different amounts of mitochondrial proteins, we were unable to detect COQ9 in patient cells, while we always detected COQ9 in controls, indicating the degradation of the truncated protein (Figure 1c).
Next, we spectrophotometrically measured several mitochondrial enzymes using mitochondria-enriched fractions of fibroblasts. To determine respiratory chain complex II/III activity we measured the reduction of cytochrome c. For complex IV activity we followed the oxidation of reduced cytochrome c, and the activity of citrate synthase, a mitochondrial matrix enzymatic marker, was measured by following the reduction of 5,5′-dithiobis-(2-nitrobenzoic acid). In line with a defect in CoQ10 biosynthesis and the defect found in fibroblasts, patient cells showed a significant decrease of complex II/III activity (Figure 2a).

(a) Biochemical measurement of activities of complex II/III, complex IV of the respiratory chain and citrate synthase. Patient fibroblasts show a significant decrease of complex II/III activity. After re-expression of wild-type cDNA in patient fibroblasts, a significant increase in complex II/III activity relative to protein, to citrate synthase as well as to complex IV activity is observed. The activity of complex II/III remained unchanged in control cells. In addition, lentiviral expression of wild-type COQ9 cDNA in fibroblasts of a patient with a disease-causing homozygous variant in COQ2 showed no effect on the activity of complex II/III, demonstrating the specificity of our complementation strategy in the COQ9 patient. (b) Total amount of CoQ10 was measured before and after lentiviral transduction as well as after treatment with CoQ10. Both patient cell lines showed significantly reduced amounts of CoQ10. After transduction patient fibroblasts with a variant in COQ9 showed a significant (about threefold) increase of CoQ10, whereas in control cells and in patient cells suffering from COQ2 deficiency no significant difference could be observed. Treatment with CoQ10 leads to an increase of the total amount of CoQ10 in both patient cell lines. (c) MS/MS chromatograms of fibroblast lysate samples: (1) detected CoQ10 in a control; (2) 6-DMQ10 in a control is not present; (3) low detection of CoQ10 in a COQ9 patient; (4) detection of 6-DMQ10 metabolite in a COQ9 patient. (d) Quantification of the 6-DMQ10 levels in COQ9-mutant fibroblasts before and after viral complementation with wild-type COQ9. The amount of 6-DMQ10 is significantly reduced in COQ9-mutant fibroblasts after transduction. (e) Treatment with CoQ10 in control and patient fibroblasts. Supplementation rescues complex II/III activity in fibroblasts derived from COQ9 and COQ2 patients whereas no differences were measured in control fibroblasts. Statistics: ***P<0.001, **P<0.01 and *P<0.05 relative to control or the indicated condition. All experimental data were obtained in at least three independent experiments.
To investigate whether the COQ9 variant is causal for the observed cellular phenotypes, we expressed the functional protein using a lentiviral expression vector containing COQ9 wild-type cDNA. Expression of COQ9 was confirmed by western blotting (Figure 1c).10
The expression of COQ9 in control cells with normal or reduced CoQ10 levels due to COQ2 variants had no influence on complex II/III activity (Figure 2a). However, after expression of wild-type cDNA in patient fibroblasts carrying the COQ9 variant, we observed a significant fourfold increase in complex II/III activity.
As COQ9 is involved in the biosynthesis of CoQ10, the total amount of CoQ10 was measured using ultra-performance liquid chromatography coupled to tandem mass spectrometry (UPLC–MS/MS). Both CoQ10 patient cell lines showed significantly reduced amounts of CoQ10, with 10 pmol/mg protein in fibroblasts with COQ2 variants and 7.3 pmol/mg protein in fibroblasts with the COQ9 variant (normal range 46.5–90.5 pmol/mg protein). After transduction, fibroblasts with mutant COQ9 showed a significant (about threefold) increase of CoQ10 levels, whereas the CoQ10 levels remained unchanged in the COQ2 mutant cells (Figure 2b). These findings provide evidence for a causal relation of the identified COQ9 variant, the CoQ10 deficiency and the reduced complex II/III activity.
Investigations in a mouse model and recent human studies demonstrated that COQ9 is a lipid-binding protein, which has structural homology to the TFR family (bacterial transcriptional regulators).9, 11 However, Lohman et al.9 demonstrated that COQ9 is unlikely to act as a transcription factor and suggested that the TFR structural fold has been repurposed during evolution. Instead of catalyzing a specific reaction of the biosynthesis of CoQ10 by itself, it has been speculated that COQ9 binds either CoQ10 or a CoQ10 precursor and forms a complex with COQ7. COQ7 catalyzes the hydroxylation of 6-demethoxy CoQ10, which constitutes the pre-final step in CoQ biosynthesis.9 The clearly reduced amount of COQ7 in the patient cell line, which is rescued by increasing COQ9, indicates a COQ7-stabilizing function of COQ9 (Figure 1c).
Of note, in COQ9-mutant mouse as well as human fibroblasts a potential intermediate of the CoQ10 biosynthesis has been described.6, 11 In mice, this metabolite was identified as 6-demethoxy ubiquinone9. Detailed human studies are lacking so far. In order to characterize the metabolite in human cells, we performed different UPLC-ESI-MS/MS analysis techniques such as full scan, precursor ion scan or multiple reaction monitoring mode. In accordance with the studies in mice we specifically detected 6-demethoxy ubiquinone10 in COQ9 fibroblasts (Figure 2c and d).As 6-demethoxy ubiquinone10 is hydroxylated by COQ7 to produce 6-hydroxy ubiquinone10, the accumulation of this intermediate can be explained by the reduced amount of COQ7.9 As expected, the expression of COQ9 normalized COQ7 and 6-demethoxy ubiquinone10 levels (Figure 2d). The metabolite was not detected in other cell lines.
CoQ10 supplementation has been reported to be beneficial in some CoQ10-deficient patients. To test the treatment effect in our cell lines we supplemented them with 5 μM CoQ10 for 1 week according to Lopez et al.12 Treatment with CoQ10 rescued complex II/III activity in COQ9-mutant fibroblasts significantly to ~80% of the lowest control value (Figure 2e). Nearly the same effect was observed in COQ2-mutant fibroblasts, whereas no differences were measured in control fibroblasts. The content of CoQ10 in patient fibroblasts markedly increased up to about 11-fold after treatment, reaching control values (Figure 2b). Interestingly, in patient fibroblasts with a genetic defect in COQ9 no 6-demethoxy ubiquinone10 was detected after treatment with CoQ10, which might be due to a reduced endogenous biosynthesis by sufficient exogenous supplementation with CoQ10 (Figure 2d).
In conclusion, our study provides additional insights into the clinical and cellular consequences of COQ9 deficiency. The severe clinical course observed in our patient suggests a critical function of COQ9 already during prenatal development. The case, together with the description of Duncan et al., places COQ9 deficiency among the most severe forms of CoQ10 metabolism disorders, comparable to descriptions in patients with defects in COQ2 or COQ4.6, 13, 14 However, further clinical descriptions will be required to evaluate the specific clinical spectrum of patients with COQ9 mutations. As suggested by Duncan et al.,6 CoQ10 treatment might be helpful in COQ9-deficient children. Accordingly, in unclear cases of neonatal/early childhood mitochondrial disease, an empirical medication with CoQ10 is recommended.
References
Laredj LN, Licitra F, Puccio HM : The molecular genetics of coenzyme Q biosynthesis in health and disease. Biochimie 2014; 100: 78–87.
Bentinger M, Tekle M, Dallner G : Coenzyme Q—biosynthesis and functions. Biochem Biophys Res Commun 2010; 396: 74–79.
Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L : Genetics of coenzyme q10 deficiency. Mol Syndromol 2014; 5: 156–162.
Emmanuele V, López LC, Berardo A et al: Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol 2012; 69: 978–983.
Quinzii CM, Emmanuele V, Hirano M 2014 Clinical presentations of coenzyme q10 deficiency syndrome. Mol Syndromol 2014; 5: 141–146.
Duncan AJ, Bitner-Glindzicz M, Meunier B et al: A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet 2009; 84: 558–566.
Haack TB, Gorza M, Danhauser K et al: Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab 2014; 111: 342–352.
Elstner M, Andreoli C, Klopstock T, Meitinger T, Prokisch H : The mitochondrial proteome database: MitoP2. Methods Enzymol 2009; 457: 3–20.
Lohman DC, Forouhar F, Beebe ET et al: Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci USA 2014; 111: E4697–E4705.
Haack TB, Kopajtich R, Freisinger P et al: ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet 2013; 93: 211–223.
García-Corzo L, Luna-Sánchez M, Doerrier C et al: Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum Mol Genet 2013; 22: 1233–1248.
López LC, Quinzii CM, Area E et al: Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PloS One 2010; 5: e11897.
Jakobs BS, van den Heuvel LP, Smeets RJ et al: A novel mutation in COQ2 leading to fatal infantile multisystem disease. J Neurol Sci 2013; 326: 24–28.
Brea-Calvo G, Haack TB, Karall D et al: COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet 2015; 96: 309–317.
Acknowledgements
This project was supported by the BMBF-funded German Network for Mitochondrial Disorders (mitoNET #01GM1113C) and by the E-Rare project GENOMIT (01GM1207). TBH was supported by the BMBF through the Juniorverbund in der Systemmedizin ‘mitOmics’ (FKZ 01ZX1405C).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website .
Supplementary information
Rights and permissions
About this article

Cite this article
Danhauser, K., Herebian, D., Haack, T. et al. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur J Hum Genet 24, 450–454 (2016). https://doi.org/10.1038/ejhg.2015.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.133
This article is cited by
-
An ETFDH-driven metabolon supports OXPHOS efficiency in skeletal muscle by regulating coenzyme Q homeostasis
Nature Metabolism (2024)
-
Mutations of GEMIN5 are associated with coenzyme Q10 deficiency: long-term follow-up after treatment
European Journal of Human Genetics (2024)
-
Mendelian gene identification through mouse embryo viability screening
Genome Medicine (2022)
-
A family segregating lethal neonatal coenzyme Q10 deficiency caused by mutations in COQ9
Journal of Inherited Metabolic Disease (2018)
