
Abstract
Defects in complex II of the mitochondrial respiratory chain are a rare cause of mitochondrial disorders. Underlying autosomal-recessive genetic defects are found in most of the ‘SDHx’ genes encoding complex II (SDHA, SDHB, SDHC, and SDHD) and its assembly factors. Interestingly, SDHx genes also function as tumor suppressor genes in hereditary paragangliomas, pheochromocytomas, and gastrointestinal stromal tumors. In these cases, the affected patients are carrier of a heterozygeous SDHx germline mutation. Until now, mutations in SDHx associated with mitochondrial disease have not been reported in association with hereditary tumors and vice versa. Here, we characterize four patients with isolated complex II deficiency caused by mutations in SDHA presenting with multisystem mitochondrial disease including Leigh syndrome (LS) and/or leukodystrophy. Molecular genetic analysis revealed three novel mutations in SDHA. Two mutations (c.64-2A>G and c.1065-3C>A) affect mRNA splicing and result in loss of protein expression. These are the first mutations described affecting SDHA splicing. For the third new mutation, c.565T>G, we show that it severely affects enzyme activity. Its pathogenicity was confirmed by lentiviral complementation experiments on the fibroblasts of patients carrying this mutation. It is of special interest that one of our LS patients harbored the c.91C>T (p.Arg31*) mutation that was previously only reported in association with paragangliomas and pheochromocytomas, tightening the gap between these two rare disorders. As tumor screening is recommended for SDHx mutation carriers, this should also be considered for patients with mitochondrial disorders and their family members.
Similar content being viewed by others


Introduction
Mitochondrial ATP production is coupled to the oxidation of the electron carriers NADH and FADH2. This oxidative phosphorylation system (OXPHOS) consists of four complexes (complex I through complex IV) of the electron transport chain (ETC) together with ATP synthase (complex V). Defects in these large protein complexes, which are encoded by both the nuclear and the mitochondrial genomes, result in mitochondrial dysfunction and cause mitochondrial disease.
Mutations in complex II (CII), the smallest complex of the electron transport chain, are relatively rare among the many causes for mitochondrial disorders.1 CII consists of four nuclear-encoded subunits (succinate dehydrogenase subunits A to D) and is located at the mitochondrial inner membrane. It has a function in the citric acid cycle, where it oxidizes succinate to fumarate, as well as in the mitochondrial ETC by transferring electrons to ubiquinone. CII is the only oxidative complex of the respiratory chain that does not contribute to the proton gradient. The largest catalytic subunit, the flavoprotein SDHA, oxidizes succinate with a coupled reduction of the cofactor FAD. SDHB, the other catalytic subunit, then mediates electron transfer to ubiquinone. Further structural components of CII are two anchoring subunits, SDHC and SDHD.
Tissues with high energy demand such as the heart, skeletal muscles, and the (central) nervous system are most vulnerable to mitochondrial dysfunction. This explains the typical findings in patients with mitochondrial disorders, such as (cardio)myopathy, optic atrophy, leukoencephalopathy, or Leigh syndrome (LS, MIM 256000). LS is an early-onset progressive neurodegenerative disorder with a characteristic neuropathology consisting of focal, bilateral lesions in one or more areas of the central nervous system, including the basal ganglia, thalamus, cerebellum, and spinal cord that are detected on cerebral MRI. The underlying genetic defects are heterogeneous,2 often a deficiency of complex IV or cytochrome C oxidase (COX), the terminal enzyme of the mitochondrial respiratory chain, is causative.3
Until now, 11 different autosomal-recessive pathogenic mutations in SDHA have been described that caused mitochondrial disorders in a total of 25 patients. All patients had isolated CII deficiency in examined tissues. In nine of the patients, the clinical phenotype consisted of LS on cerebral MRI, sometimes combined with leukodystrophy and different multisystem involvement. Most of these patients showed a regression starting in the first year of life accompanied by one or more of the following signs and symptoms: ataxia, dystonia, myopathy, optic atrophy, hepatomegaly, seizures, lactic academia, and elevated citric acid cycle intermediates in urine.4, 5, 6, 7, 8, 9, 10, 11 In addition, a Bedouin family with 15 members having isolated dilated cardiomyopathy has been described,12 and a single patient with cardiomyopathy and leukodystrophy.13
A mutation in the CII assembly factor SDHAF1 (see Ghezzi et al14) and recently also in SDHB13 and SDHD15 have also been found to cause LS or LS-like symptoms, although so far LS was not found in association with mutations in SDHC.
Mutations in SDHA and the other SDHx genes do not only influence OXPHOS but also cause a cascade of molecular events leading to, among others, the abnormal stabilization of hypoxia-inducible factors (HIFs), thereby promoting cell proliferation, angiogenesis, and tumorigenesis.16 Mutations in SDHA have also been found in hereditary paragangliomas (PGLs) and pheochromocytomas (PHEOs),17, 18, 19, 20 a specific subtype of gastrointestinal stromal tumors (GISTs),21, 22, 23, 24, 25, 26, 27, 28 and very rarely in pituitary adenoma.18
Patients with mitochondrial disorders due to mutations in SDHA harbor homozygeous or compound heterozygeous germline mutations in SDHA, causing mitochondrial dysfunction in every single cell of their body. These mutations segregate in the family in an autosomal-recessive mode, implicating that parents as well as siblings of the patients or their parents can have carrier status. Patients with the described hereditary tumors also harbor heterozygeous germline mutations in SDHA. Only a ‘second (genetic) hit’ will turn a healthy cell into a tumor cell. This general principle of tumor biology is known as Knudson hypothesis29 and has been proven for PGLs, PHEOs and GISTs due to SDHA mutations.21, 23
In the described tumors also, mutations in the other genes encoding CII subunits as well as SDHAF2 have been found, but not in genes encoding other OXPHOS complexes. Other genetic defects causing hereditary PGL/PHEO include the RET proto-oncogene, the NF1 tumor suppressor, and the VHL tumor suppressor gene (reviewed in Gimenez-Roqueplo et al30). The most common genetic alterations in GISTs are activating mutations in the receptor tyrosine kinases KIT and PDGFRA;31 more recently, mutations in BRAF and NF-1 have also been documented.23
In this paper, we describe four patients with an isolated CII deficiency in fibroblasts and, if available, muscle, clinically causing a multisystem disorder with LS and/or leukodystrophy on cerebral MRI. Sequence analysis revealed three novel mutations in SDHA as well as three previously described mutations. Two of these novel mutations are located in intronic sequences and affect SDHA mRNA splicing. This is the first report of SDHA mutations that affect mRNA splicing. For the first time we describe the mutation c.91C>T previously found in association with PGLs and GISTs in a patient with a mitochondrial disorder.
Patients and methods
Patients
The study has been carried out in the Netherlands in accordance with the applicable rules concerning the review of research ethics committees (Commissie Mensgebonden Onderzoek Regio Arnhem-Nijmegen) and informed consent.
The patient details are summarized in Supplementary Table S1. None of the patients had PGLs, PHEs or GISTs, but patients were not specifically screened for these entities.
Molecular genetic analysis
Total RNA from blood or cultured fibroblasts was extracted using RNAbee (AMS Biotechnology, Abingdon, UK) and transcribed into cDNA using Superscript II and random primers (Invitrogen, Breda, The Netherlands). The SDHA ORF (see NM_004168.2 for reference sequence and exon numbering) was amplified into four overlapping fragments using AmpliTaq Gold 360 Master Mix (Life Technologies, Bleiswijk, The Netherlands) and exon-spanning primers. The PCR fragments were sequenced using the BigDye Terminator Kit v1.1 (Life Technologies) on a 3130xL Genetic Analyzer with M13 primers. To detect mutations on genomic DNA, total DNA was extracted using the QIAamp DNA kit (Qiagen, Venlo, The Netherlands), and the proper exons were PCR amplified with primers carefully chosen not to amplify the pseudogenes and sequenced with these same primers. Primer sequences for both cDNA and genomic DNA are available as Supplementary Data. Mutations have been submitted to the LOVD2 database32 at http://chromium.liacs.nl/LOVD2/SDH/home.php.
Sequence alignment with SDHA orthologs was done using the EMBL-EBI Clustal Omega webtool at https://www.ebi.ac.uk/Tools/msa/clustalo/.
Cell culture
Fibroblasts were cultured using standard procedures in M199 medium (Gibco, Life Technologies) supplemented with 10% fetal calf serum (FCS, GE Healthcare, Diegem, Belgium), and 1% penicillin/streptomycin (Gibco) at 37 °C with 5% CO2. The 293FT cells (Invitrogen) were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/l glucose, 10% FCS, 4 mM L-glutamine, 1 mM MEM sodium pyruvate, 0.1 mM MEM nonessential amino acids, 1% penicillin/streptomycin, and 500 μg/ml geneticin (G418). During the transfections for lentivirus production, the medium did not contain penicillin/streptomycin and geneticin (according to Invitrogen recommendations). Cell lines used were the four patient lines described here, as well as the control fibroblast line (C) and two complex I (CI)-deficient cell lines P5 (homozygous NDUFS7c.364G>A (p.Val122Met)) and P6 (NDUFS7 c.364G>A (p.Val122Met) and c.449T>C (p.Met150Thr)).
Enzyme activity measurements
The activities of the separate respiratory chain complexes, citrate synthase (CS), and total protein in the fibroblasts and muscle biopsies were measured spectrophotometrically as described before33, 34 as part of our routine diagnostics. Measurements were only accepted when each of the duplicate values was within a 10% range of their average.
Fibroblast enzyme activities were corrected over total protein, and muscle enzyme activities were corrected over CS.
Assays of mitochondrial energy-generating system capacity
Semiconfluent fibroblast cultures (total of ∼450 cm2) were harvested by trypsinization. Cells were washed in phosphate-buffered saline (PBS) with 10% FCS and resuspended in 200 μl PBS/10% FCS.
Incubations were performed essentially as described previously35 in 50 μl (100 μl in the case of ATP production) reactions in membrane screw caps tubes with, in the case of the 14C-substrate oxidations, an insert vial containing hyamine. All assays contained 30 mM potassium phosphate pH 7.4, 75 mM potassium chloride, 8 mM Tris, 1.6 mM EDTA, 5 mM MgCl2, 0.04 mg/ml digitonin, 0.2 mmol/l p1,p5-di(adenosine-5′) pentaphosphate (myo-adenylate kinase inhibitor), 2.0 mM ADP, and 20 mM creatine. For the oxidation rate and ATP production from pyruvate, the assays were supplemented with 1 mM pyruvate and 1 mM malate, with or without 3.73 kBq of [1-14C]pyruvate. Similarly, for similar reaction from succinate, the assays were complemented with 1 mM succinate and 5 mM acetyl-L-carnitine, with or without 3.73 kBq of [1,4-14C]succinate. ATP production was measured in the presence and absence of 2 mM arsenite (when pyruvate+malate were the substrates) or 4 μg/ml rotenone and 4 μg/ml antimycin A (when succinate+acetyl-carnitine were the substrates). The inhibited rates were subtracted from the uninhibited rates. The assays were started by the addition of 5 μl (10 μl for the ATP production) of the cell suspension and stopped after 20 min in a shaking water bath at 37 °C by the addition of perchloric acid. Following incubation, the reaction mixtures were processed either for measurement of the released 14CO2 or the quantification of ATP, both as previously described.35 An aliquot of the cell suspension was used for CS measurements, to be used for normalization.
Lentiviral complementation
A gateway compatible expression vector for wild-type SDHA with a stop codon before the gateway cassette was purchased from Source BioScience (Nottingham, UK) and recombined with the pLenti6.2V5-DEST destination vector (Invitrogen) using the Gateway LR Clonase II Enzyme Mix (Invitrogen) according to the manufacturer’s instructions. The resulting pLenti6.2-V5-SDHA or the control construct pLenti6.2V5-AcGFP (Aequorea coerulescens Green Fluorescent Protein) was transfected into 293FT cells together with a packaging mix consisting of pLP1, pLP2, and pLP/VSVG using Lipofectamine 2000 (Invitrogen). At 24 h after transfection, the medium was refreshed, and at 72 h after transduction, supernatants containing the viral particles were harvested. The supernatants were cleared by centrifugation (5 min, 300 g) and the viral particles were stored at −80 °C before use. Infections were performed on fibroblasts in 25 cm2 flasks with 1 ml of virus containing supernatant in the presence of 6 μg/ml polybrene (Sigma, Zwijndrecht, The Netherlands). At 24 h after infection, the medium was refreshed, and 48 h after transduction, the selection medium was added (M199, 20% FCS, penicillin/streptomycin (respectively 100 U/ml and 100 μg/ml), and 2 μg/ml blasticidin (InvivoGen, Toulouse, France)). Cells were selected for 14 days, in which time the mock-infected cells (without virus) died. Blasticidin-resistant cells were used for biochemical analysis within six passages after transduction.
Lysates, electrophoresis, and western blotting
Fibroblasts cell pellets were processed for mitochondrial complexes by Blue Native (BN) PAGE as described previously.36 Alternatively, total cell extracts were made by extraction in lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 10% glycerol, and 1% Triton X-100) for 10 min on ice and subsequent clearance (10 min, 13 000 g). Protein concentrations were measured using a Micro BCA protein assay kit (Thermo Scientific, Breda, The Netherlands). Then, 40 μg of the mitochondrial extracts was separated on 5–15% BN-PAGE gels,36 and 40 μg of the total extracts was separated on 7.5% SDS-PAGE.
Antisera used were: anti-V5 (Invitrogen) and anti-NDUFA9 for CI (MitoSciences, Eugene, OR, USA), anti-SDHA (MitoSciences) and anti-SDHB (Ab84622, AbCam, Cambridge, UK) for CII, anti-subunit core 2 for CIII, anti-MTCO1 (MitoSciences) for CIV, and anti-ATP5A (MitoSciences) for CV.
Structural analysis
The structure of the porcine SDHA protein was used to study the structural details of the C189G mutation. The porcine SDHA structure was solved by X-ray crystallization (PDB-file 1ZOY (http://www.wwpdb.org/), 96% identity) and was visualized using the WHAT IF (http://swift.cmbi.ru.nl/whatif/) and YASARA (http://www.yasara.org/) Twinset.37 The figures were rendered with PovRay (http://www.povray.org/).
Results
Genetic findings and their consequence for SDHA and CII expression in fibroblasts
Mutational analysis of the four patients resulted in the identification of three novel mutations in the SDHA gene (Table 1). Two of the novel mutations, as well as a previously found mutation, were predicted to affect RNA splicing, and were therefore further studied by mRNA sequencing (Supplementary Figure S1) that confirmed that SDHA splicing was abnormal in these patients. The effects of the mutations on the SDHA protein expression levels in patient fibroblast cell lines were analyzed by SDS-polyacrylamide gel electrophoresis and western blot analysis. As shown in Figure 1a, all four patient had detectable but clearly decreased levels of SDHA protein as compared with an unaffected control (C1).

Western blot analysis of SDHA and CII in patient fibroblasts. (a) Lysates of fibroblasts of patients with SDHA mutations (P1–4) and a control cell line (C) were separated by SDS-PAGE and subsequently blotted to PVDF membrane. Membranes were probed with anti-SDHA and anti-actin as a loading control. (b) Blue native analysis of respiratory chain complexes in mitochondrial fractions from SDHA patient fibroblasts (P1–4) compared with control (C) and CI-deficient (P5 and P6) fibroblasts. Blots were incubated with the indicated antisera.
In patient P1, a compound heterozygote genotype was found. The paternal allele contained the c.356G>A mutation, encoding a premature stop in exon 4 (p. Trp119*) that results in nonsense-mediated mRNA decay (NMD). The maternal allele contained mutation c.248C>T. This patient has been previously described by Horváth et al,4 but in contrast to their conclusion that the c.248C>T mutation would result in a missense mutation p.Ala83Val, we found that this mutation introduces a novel splice acceptor site in exon 3 that results in an in-frame deletion of the last 66 nucleotides of exon 3 (r.247_312 del). RT-PCR and Sanger sequencing revealed two fragments, with the majority of the signal coming from the r.247_312del fragment, in addition to detectable amounts of the normal fragment size (Supplementary Figure S1). As the paternal allele encodes a premature stop codon, the residual amount of full length protein as seen by western blot analysis (Figure 1) can only result from partially correctly spliced mRNA from the maternal allele. The p.Ala83_Gln104del protein predicted from the incorrectly spliced mRNA was not detected, indicating that this protein is unstable.
Patient P2 was also found to carry compound heterozygous mutations. The maternal allele carried a c.91C>T mutation, resulting in a stop codon at position 31 of the protein (p.Arg31*). This mutation has not been observed before in Leigh disease patients. Interestingly, it has been previously reported as a germline mutation in several Dutch pheochromocytoma–paraganglioma syndrome patients19 and also in GISTs,21, 23 and has been found to result in NMD from this allele. The paternal allele of patient P2 contained a novel missense mutation (c.565T>G, p.Cys189Gly). Cys189 is the first of three adjacent conserved cysteine residues located in the N-terminal subdomain of the FAD binding domain of SDHA.38 This patient had the highest residual SDHA protein expression and CII assembly levels in our group of patients as seen in Figure 1. All (or nearly all, in case of incomplete NMD) remaining protein will carry the Cys189Gly mutation.
Patient P3 was found to carry a novel homozygous c.1065-3C>A mutation. Both parents were heterozygous carriers of the mutation. This mutation of the third from the last nucleotide of intron 8 affects the splice acceptor site and results in skipping of exon 9 with a consequential shift in the reading frame. Most of the incorrectly spliced mRNA will therefore be degraded because of NMD. RT-PCR and Sanger sequencing showed two fragments, including the normal size fragment, indicating that there is some residual normal splicing of SDHA mRNA that explains the presence of a small amount of residual SDHA protein, as seen in Figure 1a.
Patient P4 contained a novel homozygous mutation of the penultimate nucleotide of the first intron (c.64-2A>G). The mutation was observed to be heterozygous in both parents. Sequencing of the mRNA fragments revealed two fragments. A minor fragment resulted from an alternative splice acceptor site at position r.73 in exon 2, resulting in a deletion of 10 nucleotides (r.64_73del) and therefore creating a nonsense mRNA that will be degraded and not yield protein. The predominant fragment resulted from an alternative splice acceptor site in the first intron at position c.64-54, resulting in an insertion of r.64-54_64-1;64-2a>g into the mRNA. As these 54 nucleotides do not introduce a frameshift and do not contain internal stop codons, the size of the predicted protein is increased with 18 amino acids. Whether the 18 extra amino acids affect the SDHA protein is unclear, but is seems that there is some stable protein detected on western blot. The expected change in molecular weight is likely too small to be detectable by western blot analysis.
In all patients, the decreased levels of SDHA protein resulted in decreased levels of the holocomplex II, as established by BN electrophoresis of the intact respiratory chain complexes followed by western blot analysis of the different complexes (Figure 1b). The reduced levels of SDHA in the patients resulted in reduced incorporation of SDHA into the holocomplex II and also in less holocomplex formation as indicated by the reduced levels of SDHB at the position of CII in the gel. The amounts of all other ETC complexes were found to be unaffected, although it seems that the amount of CIV is slightly increased in the SDHA-deficient lines as compared with the control and CI-deficient lines.
Biochemical analysis of CII-deficient muscle samples and fibroblasts
From two of the patients (P2 and P4), muscle biopsies were available, and they were subjected to routine diagnostics measurements including the overall activity of the mitochondrial energy-generating system.35 The results are included in Supplementary Table S1. We noted slightly decreased oxidation rates of pyruvate in the presence of either malate or carnitine. In contrast, a large decrease in oxidation rate was found when the CII substrate succinate was used. Residual activities (percentage of lowest reference value) were 13% for patient P2 and 30% for patient P4. In addition, the ATP production from pyruvate was affected quite severely in patient P2, with only 26% residual activity. Measurement of all single enzyme (complex) activities showed a clearly reduced CII activity (residual activity of 8% for P2 and 26% for P4), whereas all other activities were considered normal, although CIII was relatively low in patient P2 (60% residual activity).
The enzymatic activities of the complexes of the electron transport chain as well as complex V were analyzed in the patient’s fibroblasts. The activities were compared with a control fibroblast line (C) as well as two known CI-deficient cells (P5 and P6). As shown in Table 2, the SDHA patients showed clear CII enzyme activity deficiency in their fibroblasts. As CII is rate limiting for the SCC activity (succinate: cytochrome c oxidoreductase activity), the CII-deficient patients also display a SCC deficiency. Activities from the other ETC complexes were normal. Residual CII activity did not appear to correlate with the levels of residual SDHA protein nor with the residual CII levels; for example, P2 showed the lowest CII enzymatic activity (Table 2) with the highest residual SDHA protein levels (Figure 1a) and most CII as detected with anti-SDHA as well as with anti-SDHB (Figure 1b). This may indicate that the mutation present in the paternal allele of this patient results in a mutant (p.Cys189Gly) SDHA protein that can still be (partially) incorporated but has reduced enzymatic activity.
To corroborate our results on the single enzyme complex activities, we tested whether the effect of the SDHA mutations in these patient could also be detected in the energy-generating capacity of intact fibroblasts. Similar to the measurements performed in fresh muscle biopsies, we performed oxidations of radiolabeled substrates. The oxidation rate of pyruvate in the presence of malate was not affected in the SDHA-deficient fibroblasts as compared with control lines (Figure 2a). Similarly, the production of ATP and phospho-creatine (CrP) from pyruvate+malate was not affected (Figure 2c). In contrast, the oxidation rate of succinate in the presence of acetylcarnitine was significantly decreased (Figure 2b), and similarly the production of ATP+CrP from succinate+acetylcarnitine was affected (Figure 2d).

Energy-generating capacity of intact fibroblasts measured as CO2 and ATP production. The energy-generating capacity of fibroblasts was assayed as described in Patients and methods section using either pyruvate+malate (a, c) or succinate+acetylcarnitine (b, d) as substrates and measuring either CO2 (a, b) or ATP production (c, d). Results of unpaired t-tests with Welch’s correction are show. ns, not significant; *P=0.013; ***P=0.001.
Lentiviral complementation and structural analysis of the p. Cys189Gly mutation
As the protein levels of SDHA and the CII holocomplex in patient P2 were the highest in our CII-deficient patient cohort, but the residual enzyme activity was lower than of the other patients, we further investigated the Cys189Gly mutation.
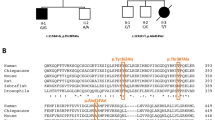
As shown in Figure 3a, the Cys189 lies in a highly conserved region of the protein and this residue is conserved from humans to drosophila. Calculations based on the degree of conservation of amino acid residues in sequence alignments derived from closely related sequences predict this mutation to be pathogenic, with a Provean score of −7.823 (‘deleterious’) and a SIFT score of 0.002 (‘damaging’). Analysis of the available porcine SDHA crystal structure (97% identical amino acid sequence to human SDHA) showed that Cys189 is located in the central part of the protein involved in FAD binding. Although the cysteine residue is not directly interacting with FAD, replacing cysteine with a glycine, which has no side chain, will have an effect on the position of the surrounding amino acids and is therefore predicted to interfere with FAD binding (Figure 3b and c).

Modeling of p.Cys189Gly in SDHA. (a) Alignment of human SDHA with orthologous proteins. Cys189 is indicated by an arrow. (b) The location of the human SDHA (gray) mutation p.Cys189Gly (red) is shown in the porcine complex II crystal structure, and the other three CII subunits are light blue. The FAD redox factor (purple) is bound to the SHDA subunit, the iron–sulfur clusters are in yellow, and the heme group in orange. (c) Detail of the proximity of the wild-type (green) cysteine to the FAD moiety. The glycine residue (red) present in the mutant protein is a smaller amino acid.
To test the pathogenicity of the Cys189Gly mutation in vitro, we introduced the wild-type SDHA cDNA into a lentiviral expression system, and patient and control fibroblasts were infected and selected for stable SDHA expression. GFP-expressing viruses were used as a control for the selection procedure. Respiratory chain enzyme activities were measured in the stably expressing cell lines and clearly showed that CII activity is partially, but significantly, restored, whereas the activity of the other enzyme complexes was unaffected by the complementation procedure (Figure 4).

Complementation of P2 fibroblasts with wild-type SDHA partially restores CII activity. Patient P2 and control (C) fibroblasts were transduced with lentiviruses carrying either the wild-type SDHA gene or GFP (green fluorescent protein) as a control. The activity of the different respiratory chain complexes and CV were analyzed in duplicate from two separate cell pellets of the stably selected cells. (a) Absolute measures of the different OXPHOS enzymes. Error bars represent SD values. (b) Efficiency of the complementation expressed as percentage of the GFP complemented controls. *P=0.02 in an unpaired t-test.
Discussion
Here we present four patients with isolated CII deficiency in fibroblasts (and muscle where available) leading to a multisystem mitochondrial disorder. We report three novel mutations and three previously described mutations in the SDHA gene.
Two of the new mutations were found in intronic sequences affecting the SDHA mRNA splicing. The novel c.1065-3C>A mutation resulted in incomplete skipping of exon 9 and the novel c.64-2A>G mutation in two different spicing products either resulting in a 18 amino acid insertion into the protein or a 10-nucleotide deletion (and therefore causing a frameshift) into the mRNA.
A previously described mutation, c.248 C>T located in exon 3, has been reported as a missense mutation resulting in alanine 83 to valine.4 However, our data indicate that the c.248C>T results in the generation of a novel splice acceptor site. At the protein level, all three splice site mutations lead to reduced SDHA expression and less CII holocomplex formation.
The third novel mutation described here, c.565T>G (p.Cys189Gly), has a different pathological mechanism. This mutation causes a single amino acid substitution that affects the SDHA protein expression and CII holocomplex formation, although considerable less than the other mutations described here. The patient carrying this heterozygous mutation also carried a premature stop codon generating mutation (p.Arg31*) on the other allele but still showed considerable SDHA protein expression levels in the fibroblast. However, remarkably little enzyme activity could be detected, indicating that in this case not merely the absence of SDHA protein induced the pathology, but instead absence of CII activity was the cause. Lentiviral complementation of the patient fibroblasts with the wild-type SDHA cDNA resulted in a significant 2.5-fold increase in CII enzymatic activity, whereas all other enzyme complex activities remained the same. Although the CII activity was only partially restored, this result proves that the patient mutation indeed causes reduced CII activity. The fact that full complementation was not obtained may have been because of the high levels of mutant protein in the cell, possibly functionally interfering with the transgenically expressed wild-type protein. From the crystal structure we noted that the affected Cys189 residue is located in close proximity of the FAD redox cofactor. The mutation to the smaller glycine residue is expected to affect the local configuration of the loops directly involved in binding the FAD.
The literature on SDHA protein expression versus CII enzyme activity is limited. However, one other mutation affecting the FAD binding has been previously reported in patients from a family affected by late-onset optic atrophy, ataxia, and a proximal myopathy.5 Surprisingly, this mutation, c.1375C>T (p.Arg408Cys), was found as a heterozygous mutation with no other mutations in the SDHA coding sequence and normal levels of both SDHA mRNA and protein. The p.Arg408Cys mutation was found to affect the specific activity of CII, likely by affecting the FAD binding.
Most interestingly, we found the c.91C>T mutation, previously described as a germline mutation associated with PGL/PHEO19 and pediatric GIST,21, 23 also causative for a multisystem mitochondrial disorder (patient P2). This severe mutation, a premature stop codon at amino acid position 31 (p.Arg31*), will abolish the SDHA protein expression. Together with the novel c.565T>G (for details see earlier), this led to a multisystem mitochondrial disorder (Leigh syndrome, epilepsy, hepatopathy, psychomotor retardation, and death by the age of 14 months).
This finding is of clinical interest for the parent carrying the c.91C>T mutation and other carriers of this, and perhaps also other, SDHA mutations. One should consider to screen family members with a carrier status of patients with mitochondrial disease for these tumors with appropriate methods (eg, plasma and urinary metanephrines, catecholamines, or functional MRI imaging).
On the other hand, one could question why patients with mitochondrial disorders due to SDHA mutations have not been reported with these kind of tumors as they should have an even more increased susceptibility. Until now, most children die before adolescence and therefore presumably die before the typical age of onset for these kind of tumors. One should definitely keep this in mind when facing patients with a milder course who reach adulthood and beyond and also consider tumor screening.
The genetic overlap of mitochondrial disorders on one hand and PGL/PHEO and GIST on the other has hereby been underlined. Mutations in SDHA, SDHAF1,14 SDHB,13 and SDHD have been implicated in both mitochondrial disorders and hereditary tumors, but the p.Arg31* is the first mutation identified to cause both disorders. Although respiratory chain disorders are among the most frequent causes of metabolic abnormalities (birth prevalence of 1/10 000),39 only a very small fraction of these are caused by mutations in the subunits of the respiratory chain complexes. Among this group, the isolated CII deficiencies are quite rare, estimated at ∼2% of all respiratory chain defects.1, 8 In addition, the contribution of the SDHA mutations to all PGL/PHEO-causing mutations is rather small, estimated at ∼3%.19 The chance of these two diseases occurring in one family is therefore rather small, but should be considered when dealing with families carrying SDHA mutation.
References
Jain-Ghai S, Cameron JM, Al Maawali A et al: Complex II deficiency-a case report and review of the literature. Am J Med Genet A 2013; 161A: 285–294.
Leigh D : Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 1951; 14: 216–221.
Pequignot MO, Dey R, Zeviani M et al: Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome C oxidase deficiency. Hum Mutat 2001; 17: 374–381.
Horvath R, Abicht A, Holinski-Feder E et al: Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry 2006; 77: 74–76.
Birch-Machin MA, Taylor RW, Cochran B, Ackrell BA, Turnbull DM : Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann Neurol 2000; 48: 330–335.
Taylor RW, Birch-Machin MA, Schaefer J et al: Deficiency of complex II of the mitochondrial respiratory chain in late-onset optic atrophy and ataxia. Ann Neurol 1996; 39: 224–232.
Bourgeron T, Rustin P, Chretien D et al: Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet 1995; 11: 144–149.
Parfait B, Chretien D, Rotig A, Marsac C, Munnich A, Rustin P : Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Genet 2000; 106: 236–243.
Van Coster R, Seneca S, Smet J et al: Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. Am J Med Genet A 2003; 120A: 13–18.
Pagnamenta AT, Hargreaves IP, Duncan AJ et al: Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol Genet Metab 2006; 89: 214–221.
Ma YY, Wu TF, Liu YP et al: Two compound frame-shift mutations in succinate dehydrogenase gene of a Chinese boy with encephalopathy. Brain Dev 2013; 36: 394–398.
Levitas A, Muhammad E, Harel G et al: Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet 2010; 18: 1160–1165.
Alston CL, Davison JE, Meloni F et al: Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J Med Genet 2012; 49: 569–577.
Ghezzi D, Goffrini P, Uziel G et al: SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet 2009; 41: 654–656.
Jackson CB, Nuoffer JM, Hahn D et al: Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J Med Genet 2013; 51: 170–175.
Bardella C, Pollard PJ, Tomlinson I : SDH mutations in cancer. Biochim Biophys Acta 2011; 1807: 1432–1443.
Burnichon N, Briere JJ, Libe R et al: SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010; 19: 3011–3020.
Dwight T, Mann K, Benn DE et al: Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab 2013; 98: E1103–E1108.
Korpershoek E, Favier J, Gaal J et al: SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab 2011; 96: E1472–E1476.
Welander J, Garvin S, Bohnmark R et al: Germline SDHA mutation detected by next-generation sequencing in a young index patient with large paraganglioma. J Clin Endocrinol Metab 2013; 98: E1379–E1380.
Pantaleo MA, Astolfi A, Indio V et al: SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011; 103: 983–987.
Pantaleo MA, Nannini M, Astolfi A, Biasco G, GIST Study Group Bologna: A distinct pediatric-type gastrointestinal stromal tumor in adults: potential role of succinate dehydrogenase subunit A mutations. Am J Surg Pathol 2011; 35: 1750–1752.
Wagner AJ, Remillard SP, Zhang YX, Doyle LA, George S, Hornick JL : Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol 2012; 26: 289–294.
Italiano A, Chen CL, Sung YS et al: SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer 2012; 12: 408.
Belinsky MG, Rink L, Flieder DB et al: Overexpression of insulin-like growth factor 1 receptor and frequent mutational inactivation of SDHA in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer 2013; 52: 214–224.
Belinsky MG, Rink L, von Mehren M : Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front Oncol 2013; 3: 117.
Miettinen M, Killian JK, Wang ZF et al: Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol 2013; 37: 234–240.
Dwight T, Benn DE, Clarkson A et al: Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol 2013; 37: 226–233.
Knudson AG Jr : Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68: 820–823.
Gimenez-Roqueplo AP, Dahia PL, Robledo M : An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 2012; 44: 328–333.
Rubin BP, Heinrich MC, Corless CL : Gastrointestinal stromal tumour. Lancet 2007; 369: 1731–1741.
Bayley JP, Devilee P, Taschner PE : The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med Genet 2005; 6: 39.
Rodenburg RJ : Biochemical diagnosis of mitochondrial disorders. J Inherit Metab Dis 2011; 34: 283–292.
Janssen AJ, Smeitink JA, van den Heuvel LP : Some practical aspects of providing a diagnostic service for respiratory chain defects. Ann Clin Biochem 2003; 40: 3–8.
Janssen AJ, Trijbels FJ, Sengers RC et al: Measurement of the energy-generating capacity of human muscle mitochondria: diagnostic procedure and application to human pathology. Clin Chem 2006; 52: 860–871.
Nijtmans LG, Henderson NS, Holt IJ : Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 2002; 26: 327–334.
Krieger E, Koraimann G, Vriend G : Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins 2002; 47: 393–402.
Sun F, Huo X, Zhai Y et al: Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005; 121: 1043–1057.
Skladal D, Halliday J, Thorburn DR : Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003; 126: 1905–1912.
Acknowledgements
We thank Anne Leenders for enzymatic analysis, and Mariël van den Brand, Antoon Janssen, and members of the DNA diagnostics, tissue culture, and muscle labs of the NCMD/LGEM for their technical expertise. Work in this study was funded by the CSBR (Centres for Systems Biology Research) initiative from NWO (no: CSBR09/013V).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Renkema, G., Wortmann, S., Smeets, R. et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet 23, 202–209 (2015). https://doi.org/10.1038/ejhg.2014.80
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.80
This article is cited by
-
Disordered-to-ordered transitions in assembly factors allow the complex II catalytic subunit to switch binding partners
Nature Communications (2024)
-
Mitochondrial heterogeneity in diseases
Signal Transduction and Targeted Therapy (2023)
-
Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency
European Journal of Human Genetics (2021)
-
A novel de novo heterozygous pathogenic variant in the SDHA gene results in childhood onset bilateral optic atrophy and cognitive impairment
Metabolic Brain Disease (2021)
-
Cardiac complex II activity is enhanced by fat and mediates greater mitochondrial oxygen consumption following hypoxic re-oxygenation
Pflügers Archiv - European Journal of Physiology (2020)

{kind=link}