
Abstract
Mutations in SUCLA2, encoding the ß-subunit of succinyl-CoA synthetase of Krebs cycle, are one cause of mitochondrial DNA depletion syndrome. Patients have been reported to have severe progressive childhood-onset encephalomyopathy, and methylmalonic aciduria, often leading to death in childhood. We studied two families, with children manifesting with slowly progressive mitochondrial encephalomyopathy, hearing impairment and transient methylmalonic aciduria, without mtDNA depletion. The other family also showed dominant inheritance of bilateral retinoblastoma, which coexisted with mitochondrial encephalomyopathy in one patient. We found a variant in SUCLA2 leading to Asp333Gly change, homozygous in one patient and compound heterozygous in one. The latter patient also carried a deletion of 13q14 of the other allele, discovered with molecular karyotyping. The deletion spanned both SUCLA2 and RB1 gene regions, leading to manifestation of both mitochondrial disease and retinoblastoma. We made a homology model for human succinyl-CoA synthetase and used it for structure–function analysis of all reported pathogenic mutations in SUCLA2. On the basis of our model, all previously described mutations were predicted to result in decreased amounts of incorrectly assembled protein or disruption of ADP phosphorylation, explaining the severe early lethal manifestations. However, the Asp333Gly change was predicted to reduce the activity of the otherwise functional enzyme. On the basis of our findings, SUCLA2 mutations should be analyzed in patients with slowly progressive encephalomyopathy, even in the absence of methylmalonic aciduria or mitochondrial DNA depletion. In addition, an encephalomyopathy in a patient with retinoblastoma suggests mutations affecting SUCLA2.
Similar content being viewed by others


Main
Mitochondrial diseases are caused by genetic defects in nuclear or mitochondrial DNA (mtDNA) that disrupt function of the respiratory chain, compromising the synthesis of ATP. Most childhood-onset phenotypes are caused by autosomal recessive mutations in nuclear-encoded mitochondrial proteins. Mitochondrial diseases can manifest at any age, with almost any symptom, in almost any tissue, although the tissues with the largest dependence on oxidative energy supply, such as the central nervous system, sensory organs and skeletal muscle,1 are most commonly affected. The wide clinical and genetic heterogeneity with overlapping phenotypes makes the diagnostics of mitochondrial diseases challenging.2
mtDNA depletion syndrome is associated with many clinical phenotypes and has a variable genetic background. It can be caused by several nuclear genes, which typically impair mtDNA replication, repair or nucleotide synthesis.3 One of these genes is SUCLA2, encoding the β-subunit of the Krebs cycle enzyme ADP-forming succinyl-CoA synthetase (SCS-A). SCS catalyzes the reversible conversion of succinyl-CoA to succinate, accompanied by substrate-level phosphorylation of ADP or GDP.4 The enzyme is a heterodimer composed of a catalytic α-subunit, encoded by SUCLG1 and a β-subunit that determines the enzymes' substrate specificity for either ADP (SUCLA2) or GDP (SUCLG2). SCS is widely expressed in mammalian tissues, with predominance of either the ADP- or GDP-forming form in each tissue. SUCLG1 is ubiquitously expressed, whereas expression of SUCLA2 dominates in catabolic tissues, in which the main source of energy is ATP, such as the brain, and is induced in heart and skeletal muscle.4, 5 Patients with SUCLA2 mutations typically have progressive childhood-onset Leigh-like encephalomyopathy associated with dystonia, hypotonia, sensorineural hearing deficit, lesions of the basal ganglia, depletion of mtDNA and methylmalonic aciduria.3, 6 Over 20 patients and five different mutations in SUCLA2 have been described.6, 7, 8, 9, 10
We report here molecular basis of mitochondrial encephalomyopathy, also combined with bilateral retinoblastoma, in patients with clinical symptoms or signs previously described in association with SUCLA2 mutations: encephalomyopathy with hearing deficit and methylmalonic aciduria.
Materials and methods
Patients
The patients were investigated at the Helsinki University Central Hospital and all samples were taken for diagnostic purposes. Informed consent was obtained from the parents and the study was approved by the Ethics Committee for Pediatrics, Adolescent Medicine, and Psychiatry of Helsinki University Central Hospital.
Patient 1 is the 2/2 child of healthy parents, with a healthy sister. He was a term baby (birth weight 4620 g, length 53 cm, head circumference 36.5 cm, Apgar score 9/10). At the age of 5 months, when first investigated for slow growth of the head circumference, he was noticed to be athetotic and his motor development was delayed. His electroencephalography and brain magnetic resonance imaging (MRI) were normal. Laboratory investigations showed slightly increased blood pyruvate (101 μmol/l, reference range 40–70 μmol/l), normal lactate (2.1 mmol/l, reference range 0.5–2.2 mmol/l) and marginally increased methylmalonic acid (MMA) in urine. At the age of 2 years he could sit up independently, stand when supported and he spoke some words. Brain MRI showed atrophy of the caudate and lentiform nuclei, and blood pyruvate and urine MMA were normal. At the age of 4 years he could stand and take a couple of steps without support and walk on his knees. His fine motor skills were at the level of a 1.5-year old. At 7 years of age posturing of the hands and feet was dystonic, deep tendon reflexes were increased and he had made slight progress on his motor skills and could walk with support. He had mild ataxia and sensorineural hearing deficit. Electroneuromyography (ENMG) showed mild sensory axonal neuropathy. The atrophy seen in the brain MRI had not progressed as compared with that at the age of 2 (Figure 1a). At 2 years of age, muscle biopsy sample showed type-II atrophy, and normal histochemical activities of cytochrome c oxidase and succinate dehydrogenase. Biochemical analysis of muscle mitochondrial respiratory chain function indicated normal activities.

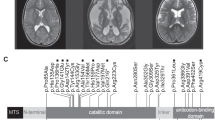
Brain axial T2-weighted magnetic resonance images showing typical abnormalities of patients with SUCLA2 mutations. (a) Image of patient 1 at 7 years showing atrophy of caudate (asterisk) and lentiform (arrow) nuclei. (b) Axial image of patient 2 at the age of 18 years showing severe atrophy of putamen (arrow) and caudate nucleus (asterisk).
Patient 2 is the 2/2 child of a healthy father, and a mother, who had a history of retinoblastoma. The patient’s sister is healthy. He was a term baby (birth weight 3780 g, length 53 cm and head circumference 36 cm). Mild hypotonia was noticed at birth. At the age of 5 months he was found to be extremely hypotonic with poor eye contact. At 6 months, bilateral retinoblastoma was diagnosed and successfully treated. He had organic aciduria (glutaric acid, MMA, 3-hydroxy-isovalerate, 4-hydroxyphenylacetate, 2-ketoglutarate and succinate) and increased lactate in blood (4.6 mmol/l) and CSF (3.4 mmol/l, reference range 0.6–2.7 mmol/l). At the age of 2.5 years he began to have athetoid movements. By the age of 4 years he could sit without support and at 7 years he had learned to walk with some support and could manage a wheelchair. Sensorineural hearing deficit and generalized epilepsy were diagnosed at the age of 10 years. He has remained seizure-free on valproate treatment. At 18 years of age he was able to walk supported in spite of marked hypotonia and dystonia and he communicated using gestures and signs. Brain MRI showed atrophy and increased signal intensity in the putamen and caudate nuclei (Figure 1b), and ENMG showed predominantly sensory axonal peripheral neuropathy, which was more severe in the upper limbs. The analysis of muscle histology at 7 months of age showed increased type-I fiber size, one ragged-red fiber and increased lipid droplets but no apparent ultrastructural abnormalities in the mitochondria. Biochemical analysis of mitochondrial respiratory chain showed partial deficiency of Complex I+III activities (9% of controls’ mean, when normalized to citrate synthase activity, as a measure of mitochondrial mass).
DNA sequence analysis
Genomic DNA was extracted from muscle tissue and peripheral venous blood samples using standard procedures. The 11 exons and exon–intron boundaries of SUCLA2 were amplified using PCR in eight fragments, using intronic primers (Supplementary Table 1). The DNA sequences were analyzed using the BigDye terminator Ready Reaction kit v 3.1 and ABI3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) and the sequences processed with the Sequencer 4.5 software (GeneCodes, Ann Arbor, MI, USA) and compared with the NCBI reference sequence NM_003850.2. Single-nucleotide detection of the c. 998A>G variant in SUCLA2 was studied using solid-phase minisequencing11 from 293 anonymous Finnish control samples. In addition, presence of the nucleotide variant was analyzed in exome data of 92 Finnish mitochondrial patients and 7308 Finnish control chromosomes from Sequencing Initiative Suomi data collection. The Sequencing Initiative Suomi (The SISu project) is an international collaboration between research groups aiming to build tools for genomic medicine. These groups are generating whole genome and whole exome sequence data from Finnish samples and provide data resources for the research community. The key groups of the project are from the Universities of Eastern Finland, Oulu and Helsinki and The Institute for Health and Welfare, Finland, Lund University, The Wellcome Trust Sanger Institute, University of Oxford, The Broad Institute of Harvard and MIT, University of Michigan, Washington University in St Louis, and University of California, Los Angeles (UCLA; http://sisu.fimm.fi). The SUCLA2 variant was submitted to the corresponding LOVD database (http://databases.lovd.nl/shared/genes/SUCLA2).
Molecular karyotyping
Molecular karyotyping (array comparative genomic hybridization) including DNA handling, hybridization to Agilent oligonucleotide-based 244 K platform and software used for copy number analysis have been described in detail elsewhere.12
Quantification of mtDNA
The mtDNA quantification was performed using real-time PCR and the data were analyzed as described previously,13 and the patient’s muscle mtDNA amount was compared with that of three age-matched controls’.
Protein structure modeling of human mitochondrial SCS-A
Protein sequences of homologs of α and β subunits of SCS-A were retrieved from the UniProt database (http://www.uniprot.org/) and aligned using the PROMALS3D Server (http://prodata.swmed.edu/promals3d/promals3d.php). Homology modeling for α and β subunits of human SCS-A was performed in SWISS-Model Server (http://swissmodel.expasy.org/) using α and β subunits of E. coli SCS-Aas templates14 (PDB id 1CQI). Building the SCS-A dimer, the structural analysis of the attained model and visualization of the results were performed using Discovery Studio v.2.5.5 (Accelrys).
Results
DNA analysis
Sequence analysis of SUCLA2 showed that patient 1 was homozygous for missense variant c. 998A>G (rs140963290) in exon 8 (Figure 2a), causing an amino-acid change from aspartic acid (Asp) to glycine (Gly) (p.(Asp333Gly)). The amino-acid change, Asp333Gly, occurred in a highly conserved area of the protein (Figure 2b). The parents were heterozygous carriers for this variant (Figure 2c). Patient 2 also appeared to be homozygous for the same c. 998A>G variant; however, only his father carried the variant. Molecular karyotyping indicated that the mother and the patient were heterozygous carriers of a 1.54-Mb deletion in chromosome 13q14, spanning seven genes, including SUCLA2 and RB1, the retinoblastoma gene (Figure 2d). Thereby, the patient was a compound heterozygote for c. 998A>G and the deletion, explaining the co-occurrence of mitochondrial encephalomyopathy and bilateral retinoblastoma in the patient, and the retinoblastoma in his mother. The patient was also found to have a 6.11-Mb deletion in chromosome 9 (Figure 2d). The father carried the c. 998A>G variant and the chromosome 9 deletion (Figure 2c) and based on him being healthy, this deletion is considered non-pathogenic.

Sequence analysis of SUCLA2 and segregation of the variant and deletions in families 1 and 2. (a) Missense mutation in exon 8 c. 998A>G occurs in patient 1 as homozygous. (b) Asp333 is highly conserved in species. (c) Segregation of the genetic defects in families 1 and 2. Mitochondrial encephalomyopathy: black shading; retinoblastoma: gray shading. (d) Deletions in chromosomes 9 and 13 of patient 2, deleted genes marked.
Carrier frequency
Six carriers of the c. 998A>G variant were found among 8078 control chromosomes, resulting in a carrier frequency of 1/673 in the Finnish population. None of the 92 patients in our mitochondrial disease cohort carried the variant. The variant was found once in 4503 North American alleles (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=140963290).
Quantification of mtDNA
Muscle mtDNA amount of patient 1 was 65% of controls’ mean and therefore did not meet the criteria of primary muscle mtDNA depletion (<40% of control’s mean). Sample from patient 2 was not available for quantification.
Structure–functional analysis of mutations
A homology model was built for the SCS-A based on homologous enzyme from E. coli. (Figure 3; PDB id 1CQI).15 The protein model indicated the organization of the SCS-A, with ATP- and succinyl-CoA-binding sites located on the opposite sides of the heterodimer (Figure 3a). We mapped all the known SUCLA2 mutations into the protein model. Asp333, encoded by c. 998A, is a part of the ‘power’ helix β as well as the interface between the subunits (Figure 3b). Asp333Gly is predicted to abolish the hydrogen bonds with amide nitrogen atoms of the residues from the α-subunit. Dimerization is unlikely to be affected because hydrophobic residues form the main contact surface14 and Asp333Gly will not perturb these interactions. As glycine is a known ‘helix breaker’16 the main effect of the Asp333Gly change is predicted to affect the configuration of the ‘power’ helix affecting the coordination of the phosphate ion, resulting in a much slower phosphorylation rate. Two Israeli patients carried a deletion causing skipping of exon 6 and part of exon 7 (Figure 3a); however, the RNA was expressed in the patients’ cells.6 Protein without this fragment is predicted to be unstable and incapable of ADP phosphorylation. All Faroese patients carried the mutation c.534+1G>A in intron 4,7, 8 which led to multiple exon skipping, resulting in complete absence of functional protein.8 Two Italian patients had a homozygous Arg284Cys change, an Italian/Romanian patient was heterozygous for Arg284Cys and Gly118Arg8 and two Iranian cousins were homozygous for a mutation encoding Asp251,10 an interaction partner of Arg284. Arg284 and Asp251 form one of the contacts critical for folding of the β-subunit and the stability of the dimer (Figure 3c) and mutations Arg284Cys and Asp251Asn are predicted to result in a less stable β-subunit. Gly118 is part of the ADP-binding site (Figure 3a and d) and Gly118Arg is predicted to alter the architecture of the active site and interfere with ADP binding, resulting in a correctly assembled but inactive protein. Ala103 is located in the same area of the protein as Gly118 (Figure 3d) and Ala103Asp mutation of two Italian patients9 is predicted to alter either ADP binding or ADP phosphorylation, resulting in an inactive protein.

Modeled structure of human mitochondrial ADP-forming succinyl-CoA synthetase with labeled pathogenic mutations (a). α subunit: light green; β subunit: cyan; catalytic His299: red; phosphohistidine loop: orange. Dashed arrow: tentative movement of the phosphohistidine loop between active sites. Decomposition of succinyl-CoA onto succinate and CoA in the catalytic α-subunit requires participation of a phosphate ion, which is coordinated by the N-termini of the two ‘power’ helixes: one from the α-subunit and the other from the β-subunit. This phosphate is utilized for phosphorylation of the catalytic His299 (His246 in E. coli) of the α-subunit located in the phosphohistidine loop.14 Phosphorylation of ADP takes place in the active site of the β-subunit. The movement of the phosphohistidine loop with phosphorylated catalytic histidine has been suggested to shuttle the phosphate between the active sites of both subunits.14, 15 The missing fragment in the case of skipping of exon 6 and part of exon 7, encompassing residues 221–267, contains Glu256, a critical residue for ADP phosphorylation.21 Location of Asp333, a part of ‘power’ helix β and the interface between subunits, within the modeled human mitochondrial SCS-A (b). Hydrogen bonds formed by Asp333: black dashed lines. Location of Arg284 and Asp251 within the modeled human SCS-A (c). Arg284 localizes to the interface between two subunits and forms a number of contacts with neighboring residues (Ser281, Asp251 and Asp151). Non-covalent bonds formed by Arg284: black dashed lines. Location of Gly118 and Ala103, in the ADP-binding site, with respect to bound ADP and the phosphohistidine loop (yellow) of subunit α of SCS-A (d).
Discussion
We show here that SUCLA2 mutations can underlie a slowly progressive childhood-onset encephalomyopathy, without mtDNA depletion and with only transient MMA increases. In addition, the close vicinity of SUCLA2 and RB1 may lead to co-occurrence of mitochondrial encephalomyopathy and bilateral retinoblastoma. Therefore, the clinical phenotype of SUCLA2 disorders spans broader than previously described.
The co-occurrence of retinoblastoma and mitochondrial encephalomyopathy was peculiar, and had been previously considered to be coincidental. However, the close vicinity of RB1 and SUCLA2 enabled them to be jointly affected by a deletion of chromosome 13q14. Retinoblastoma patients with large deletions in the 13q region have been reported to manifest with neurological symptoms such as psychomotor delay, hypotonia, deafness, seizures and brain anomalies. In a group of 42 patients with retinoblastoma and varying neurological phenotypes due to 13q deletions, 27 patients were found to have a deletion including SUCLA2.17 PCDH8 and NUFIP1 have been proposed as candidate genes for the developmental delay seen in these patients,17 but these genes were not included in the area deleted in patient 2 (Figure 2d). On the basis of our patient, we propose that SUCLA2 should be considered as one cause of neurological deficits in retinoblastoma patients with 13q14 deletions, especially in the case of psychomotor delay, motor dysfunction, sensorineural hearing deficit or increased MMA.
All previously described patients with SUCLA2 mutations presented with psychomotor delay/retardation and hypotonia, manifesting at birth or before 5 months of age, and nearly all had skeletal muscle atrophy, dystonia, sensorineural hearing deficit, basal ganglia involvement and elevated MMA in plasma or urine. Nine patients died between the ages of 8 months and 21 years due to recurrent infections. The patients, who were examined, had a respiratory chain defect and depletion of mtDNA.6, 7, 8, 9, 10 Our two Finnish patients became symptomatic at the age of 5–6 months, similar to other reported patients, but showed clearly slower progression of the disease: at the current age of 9 and 20, they are able to walk with a walker, go to school and, despite a hearing deficit and inability to speak, are able to communicate using signs and gestures. The MRI findings of patient 1 have not progressed between the age of 2 and a half years and 7 years of age. Therefore, the symptoms of the two Finnish patients were qualitatively consistent with those of previously reported patients (Table 1), despite considerably slow progression.
MMA increase in urine and mtDNA depletion have been considered typical signs of SUCLA2 disorders, even so that SUCLA2 is considered as a mtDNA depletion syndrome gene.3 MMA was only transiently increased in patient 1. Previously, three patients have been reported to have normal levels of MMA in urine.9, 10 Furthermore, the muscle mtDNA copy number in patient 1 did not fulfill the criteria of mtDNA depletion.18 These data establish that patients with normal or transiently increased levels of MMA in urine and a normal amount of mtDNA may have SUCLA2 mutations.
Our patients carried a variant in SUCLA2, changing aspartic acid 333 to glycine, previously not reported as a disease-causing variant. Several facts support the pathogenic role of this variant. Six carriers of the variant were found among 8078 Finnish control chromosomes, making the carrier frequency 1/673. The low carrier frequency in the genetically isolated Finland suggests that the variant is recently introduced into the population. The mutated amino acid is highly conserved (Figure 2b). The variant segregated with the disease in the pedigrees of the two unrelated families, as patient 1 was homozygous for the variant and his parents healthy heterozygous carriers and patient 2 was heterozygous for the variant and also carried a deletion of the same region. His father was a healthy carrier of the variant and his mother a carrier of the deletion (Figure 2c). In addition, our prediction of the effect of the variant on protein structure supported its pathogenic role.
We hypothesized that the slowly progressive phenotype of the two Finnish patients could be due to the different consequences of the Asp333Gly change on protein structure and function, as compared with previous mutations. Our structural analysis of SUCLA2 protein predicted that the previously described mutations caused either complete absence of functional protein, instability of the β-subunit leading to decreased amounts of incorrectly assembled protein, or a protein incapable of ADP phosphorylation. These effects are consistent with severe, early-onset disease. However, the Finnish Asp333Gly change was predicted to slow down the otherwise functional enzyme. The moderate effect on protein structure and function fits well with the slowly progressive disease of our patients and their mtDNA amount within normal range.
Mitochondrial dysfunction has been proposed to promote tumorigenesis and metastasis.19 We found no clear indication of increased aggressiveness of retinoblastoma in the patient with mitochondrial dysfunction, compared with his mother, suggesting that the respiratory chain defect did not affect tumor behavior.
Currently no curative treatment is available for mitochondrial diseases, except for primary ubiquinone deficiency, and the focus of therapies is on palliative care.20 After the DNA diagnosis of patient 1 was revealed, his hearing was re-assessed because patients with mutations in SUCLA2 often have a sensorineural hearing deficit. He was found to have a hearing deficit and he received a hearing aid. The finding of a genetic diagnosis may have a key role in focusing palliative care and in improving the quality life of the patient and his family.
In conclusion, our report indicates that SUCLA2 mutations should be considered in patients with psychomotor delay and motor dysfunction with hearing impairment, even if the excretion of MMA or mtDNA amounts were normal. In addition, SUCLA2 mutations/13q14 deletions should be searched for in patients with retinoblastoma and neurological deficits.
References
Skladal D, Sudmeier C, Konstantopoulou V et al: The clinical spectrum of mitochondrial disease in 75 pediatric patients. Clin Pediatr 2003; 42: 703–710.
Ylikallio E, Suomalainen A : Mechanisms of mitochondrial disease. Ann Med 2012; 44: 41–59.
Suomalainen A, Isohanni P : Mitochondrial DNA depletion syndromes - many genes, common mechanisms. Neuromuscul Disord 2010; 20: 429–437.
Johnson J, Mehus J, Tews K, Milavetz B, Lambeth D : Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes. J Biol Chem 1998; 273: 27580–27586.
Lambeth D, Tews K, Adkins S, Frohlich D, Milavetz B : Expression of two succinyl-CoA synthetases with different nucleotide specificities in mammalian tissues. J Biol Chem 2004; 279: 36621–36624.
Elpeleg O, Miller C, Hershkovitz E et al: Deficiency of the ADP-Forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet 2005; 76: 1081–1086.
Ostergaard E, Hansen FJ, Sorensen N et al: Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain 2007; 130: 853–861.
Carrozzo R, DionisiVici C, Steuerwald U et al: SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 2007; 130: 862–874.
Lamperti C, Fang M, Invernizzi F et al: A novel homozygous mutation in SUCLA2 gene identified by exome sequencing. Mol Genet Metab 2012; 107: 403–408.
Jaberi E, Chitsazian F, Ali Shahidi G et al: The novel mutation p.Asp251Asn in the β-subunit of succinate-CoA ligase causes encephalomyopathy and elevated succinylcarnitine. J Hum Genet 2013; 58: 526–530.
Suomalainen A, Syvänen A : Quantitative analysis of human DNA sequences by PCR and solid-phase minisequencing. Mol Biotechnol 2000; 15: 123–131.
Siggberg L, Ala-Mello S, Jaakkola E et al: Array CGH in molecular diagnosis of mental retardation: a study of 150 Finnish patients. Am J Med Genet 2010; 152A: 1398–1410.
Götz A, Isohanni P, Pihko H et al: Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain 2008; 131: 2841–2850.
Fraser ME, James MNG, Bridger WA, Wolodko WT : A detailed structural description of Escherichia coli succinyl-CoA synthetase. J Mol Biol 1999; 285: 1633–1653.
Joyce MA, Fraser ME, James MN, Bridger WA, Wolodko WT : ADP-binding site of Escherichia coli succinyl-CoA synthetase revealed by x-ray crystallography. Biochemistry 2000; 39: 17–25.
O'Neil KT, DeGrado WF : A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 1990; 250: 646–651.
Mitter D, Ullmann R, Muradyan A et al: Genotype–phenotype correlations in patients with retinoblastoma and interstitial 13q deletions. Eur J Hum Genet 2011; 19: 947–958.
Poulton J, Holt IJ : 163rd ENMC International Workshop: Nucleoid and nucleotide biology in syndromes of mitochondrial DNA depletion myopathy 12–14 December 2008, Naarden, The Netherlands. Neuromuscul Disord 2009; 19: 439–443.
Warburg O : On the origin of cancer cells. Science 1956; 123: 309–314.
Suomalainen A : Therapy for mitochondrial disorders: little proof, high research activity, some promise. Semin Fetal Neonatal Med 2011; 16: 236–240.
Fraser ME, Joyce MA, Ryan DG, Wolodko WT : Two glutamate residues, Glu 208 alpha and Glu 197 beta, are crucial for phosphorylation and dephosphorylation of the active-site histidine residue in succinyl-CoA synthetase. Biochemistry 2002; 41: 537–546.
Acknowledgements
We wish to thank Anu Harju and Tuula Manninen for technical assistance, Leena Valanne for neuroradiological expertise and Sequencing Initiative Suomi collaboration for the possibility to utilize their population sequence material at its early stages. This study was supported by funding from Jane and Aatos Erkko Foundation, Sigrid Jusélius Foundation, Academy of Finland, University of Helsinki (to AS) and Foundation for Pediatric Research (to PI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Matilainen, S., Isohanni, P., Euro, L. et al. Mitochondrial encephalomyopathy and retinoblastoma explained by compound heterozygosity of SUCLA2 point mutation and 13q14 deletion. Eur J Hum Genet 23, 325–330 (2015). https://doi.org/10.1038/ejhg.2014.128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.128
This article is cited by
-
Pharmacologically targetable vulnerability in prostate cancer carrying RB1-SUCLA2 deletion
Oncogene (2020)
-
SUCLA2 mutations cause global protein succinylation contributing to the pathomechanism of a hereditary mitochondrial disease
Nature Communications (2020)
-
Improving the diagnosis of cobalamin and related defects by genomic analysis, plus functional and structural assessment of novel variants
Orphanet Journal of Rare Diseases (2018)
-
Succinate‐CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients
Journal of Inherited Metabolic Disease (2016)
