
Abstract
The Forkhead box G1 (FOXG1) gene encodes a transcriptional repressor essential for early development of the telencephalon. Intragenic mutations and gene deletions leading to haploinsufficiency cause the congenital variant of Rett syndrome. We here describe Rett syndrome-like patients, three of them carrying a balanced translocation with breakpoint in the chromosome 14q12 region, and one patient having a 14q12 microdeletion excluding the FOXG1 gene. The hypothesis of long-range FOXG1-regulatory elements in this region was supported by our finding of reduced FOXG1 mRNA and protein levels in platelets and skin fibroblasts from these cases. Given that FOXG1 is not only expressed in brain but also in platelets, we have studied platelet morphology in these patients and two additional patients with FOXG1 mutations. Electron microscopy of their platelets showed some enlarged, rounder platelets with often abnormal alpha, and fewer dense granules. Platelet function studies were possible in one 14q12 translocation patient with a prolonged Ivy bleeding time and a patient with a heterozygous FOXG1 c.1248C>G mutation (p.Tyr416X). Both have a prolonged PFA-100 occlusion time with collagen and epinephrine and reduced aggregation responses to low dose of ADP and epinephrine. Dense granule ATP secretion was normal for strong agonists but absent for epinephrine. In conclusion, our study shows that by using platelets functional evidence of cis-regulatory elements in the 14q12 region result in reduced FOXG1 levels in patients’ platelets having translocations or deletions in that region. These platelet functional abnormalities deserve further investigation regarding a non-transcriptional regulatory role for FOXG1 in these anucleated cells.
Similar content being viewed by others

Introduction
The forkhead box G1 (FOXG1) gene, formerly known as Brain Factor 1 (MIM 164874), is located on chromosome 14q12, and encodes a winged-helix transcriptional repressor important for early development of the ventral telencephalon dorso-ventral patterning by integrating several signaling centres.1, 2, 3, 4 Additionally, FOXG1 controls production of specific neuronal subtypes and regulates the balance between neural progenitor cell proliferation and differentiation in the telencephalon.5, 6, 7 FOXG1 is expressed from the earliest stages of telencephalic development through adulthood. Postnatally, FOXG1 seems to be required for the survival and maturation of postmitotic neurons, and it is involved in dentate gyrus development.8 In differentiated postmitotic neurons, FOXG1 promotes cell survival through the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway and has a protective effect against the neurotoxic effect of Mecp2.9, 10
In humans, intragenic mutations and gene deletions leading to haploinsufficiency11, 12, 13, 14, 15 cause a developmental encephalopathy originally described as a congenital variant of Rett syndrome (MIM 613454) but recently redefined as the FOXG1 syndrome.16 Also, a complex chromosome 14q12 rearrangement consisting of a translocation with adjacent inversion was described in a patient with intellectual disability, brain malformations, and microcephaly.17 Similar clinical features were found in another patient with a chromosome translocation breakpoint located in the gene desert between FOXG1 and PRKD1.16 This finding, together with a similar Rett syndrome-like phenotype in patients with 14q12 deletions that do not harbor the FOXG1 gene, strongly suggests the presence of long-range regulatory elements for FOXG1 expression in this region.16, 18, 19, 20
We here report three de novo chromosome 14q12 translocations and one microdeletion in the 14q12 region in unrelated patients with microcephaly, severe intellectual disability, absent language, seizures, and corpus callosum hypoplasia. FOXG1 is a transcriptional repressor believed to be restricted to fetal and adult brain and testis.21 Given that abnormal platelet morphology and function has been described in several neuronal disorders,22 we have used platelets from these patients and two previously described FOXG1 mutation-positive patients23, 24 to study FOXG1 expression and platelet ultrastructure. We also performed functional platelet studies in two patients with either a translocation or FOXG1 mutation.
Materials and methods
Patients
Patients were referred from different clinical centres in Belgium (University Hospital Leuven and University Hospital Antwerp), Germany (University Hospital Essen, and Max Planck Institute for Molecular Genetics of Berlin) and the Netherlands (Radboud University Nijmegen). They were assessed in detail by experienced clinical geneticists and neurologists (Table 1). We obtained blood samples from all patients and skin fibroblasts from the three translocation patients with a breakpoint in chromosome 14q12. Blood from all patients was available for electron microscopy and platelet protein expression studies that do not require freshly isolated platelets. By contrast, platelet functional studies could only be performed for blood samples that reached our centre within 2–3 h after blood drawing (being patients 1 and 5 of Table 1). The Ethical committee from the University of Leuven approved these studies.
Chromosome characterization
Fluorescence in situ hybridization experiments for delineation of the 14q12 breakpoints were performed with Bacterial Artificial Chromosome clones as previously described.16 Comparative genomic hybridization (CGH) was performed using the Affymetrix SNP array 6.0 (Affymetrix, Santa Clara, CA, USA) for patients 1 and 3 and the Affymetrix 250k SNP array25 for patient 4.
Mutation analysis of FOXG1
The entire coding region of the FOXG1 gene (GenBank accession no. NM_005249.4) was sequenced on an ABI 310 automated capillary sequencer (Applied Biosystems, London, UK). Primer sequences are available on request.
Hematological counts, functional, and morphological platelet studies
EDTA anticoagulated blood was analyzed on an automated cell counter (Cell-Dyn 1300; Abbott Laboratories, Abott Park, IL, USA) to determine blood cell counts and mean platelet volume (MPV). Platelet-rich plasma (PRP) was prepared by centrifugation (15 min at 150 g) of whole blood anticoagulated with 3.8% trisodium citrate (9:1). The PRP was used for functional platelet studies (aggregation tests and ATP secretion) and for electron microscopy, as described previously.26
Isolation of nuclear extracts from fibroblasts, platelet lysates, and immunoblot analysis
Nuclear extracts from fibroblasts were obtained in a two-step lysis process at 4 °C. Cells were lysed with a first buffer (Hepes-KOH 10 mM, MgCl2 1.5 mM, KCl 10 mM, 0.5 mM DTE, 170 μM PMSF) during 10 min. After centrifugation at 300 g for 10 min, the remaining pellet was lysed with a second buffer (Hepes-KOH 10 mM, glycerol 25%, NaCl 0.4 M, MgCl2 1.5 mM, EDTA 0.2 mM, 0.5 mM DTE, 170 μ M PMSF) for 20 min. A second centrifugation step yielded the soluble nuclear extract. Total platelet protein lysates were obtained by centrifugation of the PRP at 700 g for 15 min. The secretome (platelet releasate) after stimulation of washed platelets with very strong agonists (TRAP-6 and A23187) was obtained as described previously.26 The platelet total cytosol and membrane fractions were prepared as described.27 Equal amounts of protein fibroblasts nuclear extracts (10 μg), platelet total lysates (10 or 20 μg), platelet secretome, cytosol, and membrane (20 μg) fractions were resolved by SDS-PAGE on 10% gels and transferred onto a Hybond ECL-nitrocellulose membrane (GE Healthcare, Life Sciences, Diegem, Belgium). After blocking in 5% milk powder in Tris-buffered saline-Tween 20 (10 mM Tris-HCl pH 8.0, 150 mM sodium chloride, 0.1% Tween 20) for 1 h at room temperature, the membranes were incubated overnight at 4 °C with the primary polyclonal rabbit anti-FOXG1 antibody (ab18259, Abcam, Cambridge, UK). Antibodies used as loading control were the anti-nuclear factor SP1 (sc-59, Santa-Cruz Biotechnology, Santa-Cruz, CA, USA) and anti-beta actin (#4970, Cell Signaling Technology, Leiden, The Netherlands). Membranes were then incubated for 3 h at room temperature with a HRP-conjugated secondary antibody. The subsequent staining was performed with the western blotting-enhanced chemiluminescence detection reagent (GE Healthcare, Life Sciences).
Control platelet lysates were prepared from healthy unrelated volunteers and control fibroblasts were available from two non-related patients with autoimmune thrombotic thrombocytopenic purpura after the disease crisis (for exclusion of congenital thrombotic thrombocytopenic purpura). Controls fibroblasts were age and sex matched with patients and of a similar passage number.
Quantification was performed using the Java image processing programme ImageJ 1.34 g (NIH Image software; http://rsb.info.nih.gov/ij/).
Genomic quantitative FOXG1 PCR
Deletions or duplications of the FOXG1 gene were excluded by quantitative PCR for exon 1 on genomic DNA. Quantitative PCR was performed using SYBR Green detection method and the 7500 Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). The GNAS gene was used as control gene and normalized to chromosome 1 region No.3 considered as 100%.28 Primer sequences used are available on request.
Quantitative FOXG1 mRNA expression analysis in skin fibroblasts
RNA was extracted from the patients’ fibroblasts (and from the same normal control fibroblasts as those used for immunoblot analysis) using the RNEasy Kit according to the manufacturer’s guidelines (Qiagen, Hilden, Germany) and treated with DNAse I (Roche, Mannheim, Germany) before cDNA production. cDNA conversion and qRT-PCR analysis were performed as previously described, using SYBR Green detection method and the 7500 Fast Real-Time PCR system (Applied Biosystems).29 Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as control gene for normalization (primer sequences are available on request).
Statistical analysis
Statistical differences in protein expression were evaluated using the Student’s t-test (two-tailed). A P<0.05 was chosen for levels of significance.
Results
Clinical and genetic description of patients
A detailed phenotypic overview of the patients’ clinical symptoms is presented in Table 1.
The first patient is an 8-year-old boy without dysmorphic features born at term as the only child to healthy unrelated parents with a birth weight of 4050 g (+1.3 SD) and head circumference (OFC) of 35 cm (mean). Pregnancy was uneventful. The neonatal period was characterized by feeding difficulties and excessive crying. Psychomotor delay with visual impairment based on retinal dystrophy was observed in the first year of life as were seizures. Magnetic resonance imagingof the brain showed hypoplasia of the corpus callosum. Postnatal microcephaly became evident with OFC of 46.5 cm (−4.2 SD) at the age of 8 years. At clinical examination, axial hypotonia with spastic quadriplegia and dystonic movements, being able to sit unsupported for a few minutes, as well as stereotypic hand movements with wringing were present. There was no speech, but he laughed loudly when stimulated. Chromosome analysis using fluorescence in situ hybridization revealed an apparently balanced de novo translocation: 46,XY,t(12;14)(p11;q12). Fine-mapping showed probes that hybridized proximal (RP11-125A05) and distal (RP11-667E04) to the breakpoint, with RP11-651F14 spanning the breakpoint located in the intergenic region between FOXG1 and PRKD1 (minimally 556 kb and maximally 704 kb from the 3′ end of the FOXG1 gene; Figure 1a). An array-CGH analysis showed no other chromosomal abnormalities.

(a) Physical map of the 14q12 region. Arrow heads indicate the 5′–3′ orientation of the genes in the 14q12 region. Bacterial Artificial Chromosome clones used for mapping the translocation breakpoints in patients 1–3 (P1–3) are given by small black horizontal bars. A horizontal black dashed line depicts the microdeletion identified in patient 4 (P4). Patient 5 and 6 (P5,6) carry a nonsense (p.Tyr416X) and missense (p.Ala193Thr) mutation in the FOXG1 gene, respectively. (b) Folds of expression of FOXG1 in 14q12 translocation patients (P1–3) relative to controls (C1,2) in fibroblasts. Expression of FOXG1 and housekeeping GAPDH are given as means±SD of triplicates for each cDNA sample. RQ, relative quantity; P, patient; C, control. (c) Immunoblot analysis of FOXG1 in nuclear extracts from fibroblasts (left panel) or whole protein lysates from platelets (right panel). Protein expression levels are given as mean values relative to the control samples. Error bars represent SD. Loading controls used are the nuclear transcription factor SP-1 for fibroblast’s nuclear extracts and beta-actin for platelets’ protein lysates. Statistical analysis for each patient sample was determined against controls with significance *P<0.05 and **P<0.01. P: patient; C: control.
Patient 2, a 4-year-old girl with plagiocephaly and strabismus, was born at the postmenstrual age of 35 weeks as part of a healthy non-identical twin pair. Her development was severely delayed with absent language, no sitting and poor social interaction. Seizures were noticed at the age of 6 months, with brain imaging showing callosal hypogenesis. She was noted to be progressively microcephalic (OFC of 40 cm (−4.5 SD) and 41.5 cm (−5.1 SD) at age 1 and 2 years, respectively). Chromosome analysis revealed a de novo translocation: 46,XX,t(5;14)(p13;q11.2). Fine-mapping located the breakpoint between RP11-309B23 and RP11-125A5 in a region about 200 kb distal to FOXG1 (Figure 1a).
Patient 3 is a 10-year-old girl born term from healthy parents who presented with an OFC at birth of 32 cm (−1.7 SD) and severe developmental delay with seizures since the age of 16 months. Magnetic resonance imaging showed hypogenesis of the corpus callosum. Facial dysmorphia with hypertelorism, broad nasal bridge, and thin upper lip were apparent. Dystonic movements with hand wringing were present. Severe feeding problems required tube feeding at the age of 9 years. A de novo translocation was found with breakpoint on chromosome 14q located in RP11-684G15 (Figure 1a): 46,XX,t(5;14)(p15.3;q13.1). Array-CGH analysis again showed no other chromosomal abnormalities.
No intragenic mutations in the FOXG1 gene were found in any of these translocation patients.
Patient 4 presented with profound intellectual disability, strabismus, and postnatal microcephaly. The few words she mastered at the age of 5 years were lost after onset of epilepsy. Dysmorphic features were apparent with mild midface hypoplasia, prognathism, bulbous nose tip, broad mouth with full lips, and everted lower lip. Chromosome analysis revealed a de novo ∼1.9 Mb deletion in chromosome 14 partially overlapping the breakpoints of the first three patients (28850905-30750728 according to Hg18 UCSC genome browser; SNP_A-2231981>SNP_A-2178566) × 1) (Figure 1a). The deletion includes several genes (PRKD1, G2E3, SCFD1, COCH, STRN3, AP4S1, HECTD1) but not FOXG1. This was confirmed by the finding of a heterozygous CAG repeat in the FOXG1 gene of this patient (c.218_220dup) increasing the number of glutamine repeats from 4 to 5 (p.Gln73dup). The mother of this patient carries the same duplication.
We refer to the original papers for a detailed clinical description of the FOXG1 mutation-positive patient 5 (c.1248C>G, p.Tyr416X) and patient 6 (c.577 G>A, p.Ala193Thr).23, 24 Their main clinical symptoms are, however, also listed in Table 1.
FOXG1 mRNA and protein expression in fibroblasts and platelets
Quantitative RT-PCR analysis showed a reduced FOXG1 expression in fibroblasts from all three patients with the 14q12 translocation (Figure 1b). Concordantly, immunoblot analysis of FOXG1 using fibroblast nuclear extracts showed a lower FOXG1 protein expression as compared with control samples (Figure 1c, left panel). This finding was confirmed in whole platelet protein lysates from the three translocation patients as well for the patient with 14q12 microdeletion (Figure 1c, right panel). FOXG1 protein levels were not significantly different from control samples for the patients carrying an intragenic FOXG1 mutation.
Morphological and functional platelet studies
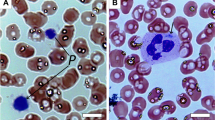
Platelet electron micrographs from the patients showed some enlarged and rather round instead of discoid platelets (Figure 2a, Table 1). Alpha granules often have a heterogeneous content and dense granules were reduced in number for patients 4–6.

(a) Platelet electron microscopy in translocation patient 2 (P 2), in patient 4 with the microdeletion (P 4), and in patient 6 with FOXG1 mutation (P 6). Platelets are more round, and alpha granules have a heterogeneous content (small black arrows) or are enlarged (small white arrows). Platelets from P4 contain pronounced microtubule-like structures (big black arrow). Bars=1 μm. (b) FOXG1 localization in normal human platelets. Immunoblot analysis using the platelet secretome (releasate), total cytosol and the remaining membranes showed mainly FOXG1 expression in the cytosol fraction that is partially released upon strong platelet activation.
Functional platelet studies could only be performed for the translocation patients 1 and 5 with the FOXG1 nonsense mutation c.1248C>G (p.Tyr416X, Table 2). Both patients never presented with a spontaneous or trauma-related bleeding tendency, but patient 1 has a mildly prolonged Ivy bleeding time. Their platelet count and size (MPV) were normal. The measurement of the MPV is probably not sensitive enough to detect some enlarged platelets as seen by electron microscopy. The PFA-100 closure time with collagen and epinephrine was prolonged for both the patients. Platelet aggregations were normal except for stimulations with a low dose of ADP and epinephrine. This type of aggregation defect is typically present in patients with a reduced platelet dense granule release.26 As expected, the ATP secretion from dense granules is absent for the epinephrine-induced platelet response for patient 1 as the aggregation response for this agonist lacks the secondary secretion-dependent phase. However, ATP secretion with both stronger agonists ADP and collagen was normal in our patients. This could mean that another intracellular amplification signaling pathway besides dense granule release is decreased in platelets from these patients as further hypothesized in the discussion. Interestingly, this is supported by the observation that immunoblot analysis using the platelet secretome, total cytosol, and the remaining membranes showed mainly FOXG1 expression in the cytosol fraction that is partially released upon strong platelet activation (Figure 2b). It is not clear whether FOXG1 is really localized in the granules or only secreted from the cytosol upon granule fusion and release during platelet activation.
Discussion
In this study, we extend findings from previous reports by describing for the first time reduced FOXG1 mRNA and protein expression in platelets and skin fibroblasts from patients having a congenital variant Rett syndrome-like phenotype with chromosomal 14q12 translocations or microdeletion that affect putative long-range cis regulatory elements for FOXG1 expression. Alterations in the expression levels of FOXG1 influence brain development. In mice, a null mutation causes early death and severe brain defects, whereas heterozygous knockout mice have learning deficits and a reduction in the size of the corpus callosum together with other patterning defects.1, 17, 30, 31, 32 On the other hand, overexpression of Foxg1 in chicken brain results in thickening of the neuroepithelium and large outgrowth of the telencephalon and mesencephalum, due to reduction of neuroepithelial apoptosis rather than increased cell proliferation.33 Similarly, in humans, intragenic mutations and gene deletions leading to haploinsufficiency11, 12, 13, 14, 15, 34, 35 are associated with the congenital variant of Rett syndrome. The importance of FOXG1 dosage during brain development is further suggested by the association of chromosome 14q12 duplications harboring FOXG1 with epilepsy, mental retardation, and severe speech impairment.36, 37, 38, 39 However, Amor et al40 recently questioned the pathogenicity of FOXG1 duplications.
Kortum et al16 described one patient with mental retardation and postnatal microcephaly who carries a 2;14 translocation with the 14q12 breakpoint mapping in a region ∼265 kb downstream of FOXG1. This finding, together with 14q12 deletions that do not harbor the FOXG1 gene but associated with a similar phenotype to that of FOXG1 mutation-positive patients, strongly suggests the presence of long-range regulatory elements of FOXG1.14, 16, 18, 19, 20 Our study further supports the existence of a long-range regulatory region. The three 14q12 translocations in this study are associated with a lower FOXG1 expression and have their breakpoint at a distance of 200 kb up to 55 6kb downstream of FOXG1 gene. In addition, the chromosomal microdeletion in patient 4 does not include the FOXG1 gene. Regulatory sequences in cis-ruption disease can be located as far as 1.3 Mb and mainly affect genes encoding developmental regulators and signaling factors (reviewed in Kleinjan and Coutinho41). Previous studies have suggested evolutionary conserved regions with gene enhancer activity located 620 and 684 kb downstream from FOXG1.16, 20 The putative region was recently narrowed down by Allou et al18 in a 0.43-Mb DNA segment encompassing the PRKD1 locus (28 945 423–29 372 834). Our data do not allow further narrowing of the regulatory region as the split signal for patient 1 was found at minimally 556 kb, whereas the study by Allou et al18 narrowed down the locus 600 kb distal to the FOXG1 coding sequence. Involvement of PRKD1 in the pathophysiology cannot be excluded though normal PRKD1 protein levels were detected in the platelets from patient 1 (data not shown). PRKD1 encodes a serine/threonine kinase involved in many cellular functions, including transport to golgi, and regulation of cell shape, motility, and adhesion.42 No human pathology associated with PRKD1 has yet been described.18
The patients we describe present clinical features that fit within the congenital variant Rett-syndrome.43 All our patients have postnatal microcephaly, severe early developmental delay with absent language, seizures, and abnormal brain morphology (callosal hypogenesis in translocation patients or delayed myelinization in the mutation-positive patients). Functional hand use was either absent or very limited in all the patients, with stereotypic movements in four out of six patients. Bruxism (4/6) and strabismus (5/6) were also fairly consistent features. In the first patient, further ophthalmic examination revealed retinal dystrophy, which is in line with the role of FOXG1 during development of the olfactory epithelium.44 In addition, we describe for the first time an association between FOXG1 haploinsufficiency and a platelet morphology and function defect. Platelets from FOXG1 haploinsufficient or mutated patients were enlarged and rounder on electron microscopy with abnormal granule morphology and/or number. Aggregations with low dose of ADP and epinephrine were reduced though the dense granule ATP secretion was normal. Our findings could indicate a role for FOXG1 in the amplification of platelet intracellular signal transduction after initial platelet activation rather than having a role in the formation and/or secretion of granules. A role for Forkhead box proteins during megakaryocyte development and platelet formation has been described for FOXO and FOXP3.45, 46 FOXP3 was shown to be important for proper megakaryopoiesis, with Foxp3-deficient mouse and human megakaryocyte progenitors having proliferation defects.45 Additionally, Foxp3sfmice were thrombocytopenic and had increased platelet volume with altered serum levels of CD40L, TXB2, and TGF-beta. One patient with a FOXP3 mutation that causes the IPEX syndrome, had defective platelet spreading and release of TGF-beta and CD40L. However, our patients had a normal platelet number and MPV, suggesting no important role for FOXG1 in megakaryopoiesis and/or platelet production. Alternatively, the presence of rounder and often larger platelets could also point to cytoskeletal defects, given the importance of the microtubule ring to maintain the characteristic platelet discoid shape.22, 47, 48 However, cytoskeletal changes typically also affect platelet secretion.26, 47 Interestingly, an important role for the PI3K/AKT pathway has been shown to stimulate FOXO3A and FOXG1 phosphorylation via epinephrine or IGF1, respectively.9, 49 The PI3K/AKT pathway is also known to be important as an amplification signaling pathway for initial platelet activation via epinephrine50 and ADP.51 Further studies are needed to define whether FOXG1 is also essential in this pathway to obtain full platelet activation. Involvement of another gene for the platelet defect cannot fully be excluded without further platelet studies that include additional patients with FOXG1 mutations.
In conclusion, this study describes four patients with de novo chromosome 14q12 defects that result in decreased expression of FOXG1 by disrupting cis-regulatory elements downstream of the gene. Additionally, this is the first study to imply a possible role for FOXG1 in platelet morphology and function.
Accession codes
References
Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E : Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 1995; 14: 1141–1152.
Danesin C, Peres JN, Johansson M et al: Integration of telencephalic Wnt and hedgehog signaling center activities by Foxg1. Dev Cell 2009; 16: 576–587.
Hanashima C, Li SC, Shen L, Lai E, Fishell G : Foxg1 suppresses early cortical cell fate. Science 2004; 303: 56–59.
Roth M, Bonev B, Lindsay J et al: FoxG1 and TLE2 act cooperatively to regulate ventral telencephalon formation. Development 2010; 137: 1553–1562.
Hanashima C, Fernandes M, Hebert JM, Fishell G : The role of Foxg1 and dorsal midline signaling in the generation of Cajal-Retzius subtypes. J Neurosci 2007; 27: 11103–11111.
Miyoshi G, Fishell G : Dynamic FoxG1 expression coordinates the integration of multipolar pyramidal neuron precursors into the cortical plate. Neuron 2012; 74: 1045–1058.
Shen Q, Wang Y, Dimos JT et al: The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci 2006; 9: 743–751.
Tian C, Gong Y, Yang Y et al: Foxg1 has an essential role in postnatal development of the dentate gyrus. J Neurosci 2012; 32: 2931–2949.
Dastidar SG, Landrieu PM, D’Mello SR : FoxG1 promotes the survival of postmitotic neurons. J Neurosci 2011; 31: 402–413.
Dastidar SG, Bardai FH, Ma C et al: Isoform-specific toxicity of Mecp2 in postmitotic neurons: suppression of neurotoxicity by FoxG1. J Neurosci 2012; 32: 2846–2855.
Ariani F, Hayek G, Rondinella D et al: FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008; 83: 89–93.
Bisgaard AM, Kirchhoff M, Tumer Z et al: Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am J Med Genet A 2006; 140: 2180–2187.
Jacob FD, Ramaswamy V, Andersen J, Bolduc FV : Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet 2009; 17: 1577–1581.
Mencarelli MA, Kleefstra T, Katzaki E et al: 14q12 Microdeletion syndrome and congenital variant of Rett syndrome. Eur J Med Genet 2009; 52: 148–152.
Papa FT, Mencarelli MA, Caselli R et al: A 3 Mb deletion in 14q12 causes severe mental retardation, mild facial dysmorphisms and Rett-like features. Am J Med Genet A 2008; 146A: 1994–1998.
Kortum F, Das S, Flindt M et al: The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet 2011; 48: 396–406.
Shoichet SA, Kunde SA, Viertel P et al: Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet 2005; 117: 536–544.
Allou L, Lambert L, Amsallem D et al: 14q12 and severe Rett-like phenotypes: new clinical insights and physical mapping of FOXG1-regulatory elements. Eur J Hum Genet 2012; 20: 1216–1223.
Santen GW, Sun Y, Gijsbers AC et al: Further delineation of the phenotype of chromosome 14q13 deletions: (positional) involvement of FOXG1 appears the main determinant of phenotype severity, with no evidence for a holoprosencephaly locus. J Med Genet 2012; 49: 366–372.
Ellaway CJ, Ho G, Bettella E et al: 14q12 microdeletions excluding FOXG1 give rise to a congenital variant Rett syndrome-like phenotype. Eur J Hum Genet 2013; 21: 522–527.
Murphy DB, Wiese S, Burfeind P et al: Human brain factor 1, a new member of the fork head gene family. Genomics 1994; 21: 551–557.
Goubau C, Jaeken J, Levtchenko EN et al: Homozygosity for aquaporin 7 G264V in three unrelated children with hyperglyceroluria and a mild platelet secretion defect. Genet Med 2012; 15: 55–63.
Bahi-Buisson N, Nectoux J, Girard B et al: Revisiting the phenotype associated with FOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics 2010; 11: 241–249.
Van der Aa N, Van den Bergh M, Ponomarenko N, Verstraete L, Ceulemans B, Storm K : Analysis of FOXG1 is highly recommended in male and female patients with Rett syndrome. Mol Syndromol 2011; 1: 290–293.
Willemsen MH, de Leeuw N, de Brouwer AP et al: Interpretation of clinical relevance of X-chromosome copy number variations identified in a large cohort of individuals with cognitive disorders and/or congenital anomalies. Eur J Med Genet 2012; 55: 586–598.
Di Michele M, Thys C, Waelkens E et al: An integrated proteomics and genomics analysis to unravel a heterogeneous platelet secretion defect. J Proteomics 2011; 74: 902–913.
Freson K, Jaeken J, Van Helvoirt M et al: Functional polymorphisms in the paternally expressed XLalphas and its cofactor ALEX decrease their mutual interaction and enhance receptor-mediated cAMP formation. Hum Mol Genet 2003; 12: 1121–1130.
Klopocki E, Schulze H, Strauss G et al: Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet 2007; 80: 232–240.
Freson K, Hashimoto H, Thys C et al: The pituitary adenylate cyclase-activating polypeptide is a physiological inhibitor of platelet activation. J Clin Invest 2004; 113: 905–912.
Eagleson KL, Schlueter McFadyen-Ketchum LJ, Ahrens ET et al: Disruption of Foxg1 expression by knock-in of cre recombinase: effects on the development of the mouse telencephalon. Neuroscience 2007; 148: 385–399.
Shen L, Nam HS, Song P, Moore H, Anderson SA : FoxG1 haploinsufficiency results in impaired neurogenesis in the postnatal hippocampus and contextual memory deficits. Hippocampus 2006; 16: 875–890.
Storm EE, Garel S, Borello U et al: Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 2006; 133: 1831–1844.
Ahlgren S, Vogt P, Bronner-Fraser M : Excess FoxG1 causes overgrowth of the neural tube. J Neurobiol 2003; 57: 337–349.
Kamnasaran D, O'Brien PC, Schuffenhauer S et al: Defining the breakpoints of proximal chromosome 14q rearrangements in nine patients using flow-sorted chromosomes. Am J Med Genet 2001; 102: 173–182.
Torgyekes E, Shanske AL, Anyane-Yeboa K et al: The proximal chromosome 14q microdeletion syndrome: delineation of the phenotype using high resolution SNP oligonucleotide microarray analysis (SOMA) and review of the literature. Am J Med Genet A 2011; 155A: 1884–1896.
Brunetti-Pierri N, Paciorkowski AR, Ciccone R et al: Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet 2011; 19: 102–107.
Yeung A, Bruno D, Scheffer IE et al: 4.45 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur J Med Genet 2009; 52: 440–442.
Tohyama J, Yamamoto T, Hosoki K et al: West syndrome associated with mosaic duplication of FOXG1 in a patient with maternal uniparental disomy of chromosome 14. Am J Med Genet A 2011; 155A: 2584–2588.
Striano P, Paravidino R, Sicca F et al: West syndrome associated with 14q12 duplications harboring FOXG1. Neurology 2011; 76: 1600–1602.
Amor DJ, Burgess T, Tan TY, Pertile MD : Questionable pathogenicity of FOXG1 duplication. Eur J Hum Genet 2012; 20: 595–596, author reply 6-7.
Kleinjan DJ, Coutinho P : Cis-ruption mechanisms: disruption of cis-regulatory control as a cause of human genetic disease. Brief Funct Genomic Proteomic 2009; 8: 317–332.
Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P : Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol 2009; 11: 545–556.
Neul JL, Kaufmann WE, Glaze DG et al: Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010; 68: 944–950.
Hatini V, Tao W, Lai E : Expression of winged helix genes, BF-1 and BF-2, define adjacent domains within the developing forebrain and retina. J Neurobiol 1994; 25: 1293–1309.
Bernard JJ, Seweryniak KE, Koniski AD et al: Foxp3 regulates megakaryopoiesis and platelet function. Arterioscler Thromb Vasc Biol 2009; 29: 1874–1882.
Cornejo MG, Mabialah V, Sykes SM et al: Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood 2011; 118: 1264–1273.
Freson K, De Vos R, Wittevrongel C et al: The TUBB1 Q43P functional polymorphism reduces the risk of cardiovascular disease in men by modulating platelet function and structure. Blood 2005; 106: 2356–2362.
Kunert S, Meyer I, Fleischhauer S et al: The microtubule modulator RanBP10 plays a critical role in regulation of platelet discoid shape and degranulation. Blood 2009; 114: 5532–5540.
Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC : Involvement of cAMP/Epac/PI3K-dependent pathway in the antiproteolytic effect of epinephrine on rat skeletal muscle. Mol Cell Endocrinol 2010; 315: 104–112.
Kim S, Garcia A, Jackson SP, Kunapuli SP : Insulin-like growth factor-1 regulates platelet activation through PI3-Kalpha isoform. Blood 2007; 110: 4206–4213.
O’Brien KA, Gartner TK, Hay N, Du X : ADP-stimulated activation of Akt during integrin outside-in signaling promotes platelet spreading by inhibiting glycogen synthase kinase-3beta. Arterioscler Thromb Vasc Biol 2012; 32: 2232–2240.
Acknowledgements
This work was supported by the ‘Excellentie financiering KU Leuven’ (EF/05/013), by research grants G.0490.10N and G.0743.09 from the Fund for Scientific Research – Flanders (FWO-Vlaanderen, Belgium), GOA/2009/13 from the Research Council of the University of Leuven (Onderzoeksraad KU Leuven, Belgium). CG has a research grant of the ‘Stichting Marguerite-Marie Delacroix’ (Belgium), and ‘Heimburger award’ (Germany). GB and CVG are Senior Clinical Investigators of the FWO – Vlaanderen. CVG is holder of the Bayer and Norbert Heimburger (CSL Behring) Chairs. This work was also supported by grants from the Consortium ‘Stronger on your own feet’ to TK and MHW and The Netherlands Organization for Health Research (ZonMw grants 990700365 to TK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Goubau, C., Devriendt, K., Van der Aa, N. et al. Platelet defects in congenital variant of Rett syndrome patients with FOXG1 mutations or reduced expression due to a position effect at 14q12. Eur J Hum Genet 21, 1349–1355 (2013). https://doi.org/10.1038/ejhg.2013.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.86
Keywords
This article is cited by
-
Diagnosis of FOXG1 syndrome caused by recurrent balanced chromosomal rearrangements: case study and literature review
Molecular Cytogenetics (2020)
-
Precisely controlling endogenous protein dosage in hPSCs and derivatives to model FOXG1 syndrome
Nature Communications (2019)
-
Regulatory variants of FOXG1 in the context of its topological domain organisation
European Journal of Human Genetics (2018)
-
RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome
Molecular Autism (2017)
-
Platelet studies in autism spectrum disorder patients and first-degree relatives
Molecular Autism (2015)
