
Abstract
We report on a young woman admitted to our Cardiology Unit because of an episode of cardiac arrest related to a long-QT syndrome (LQTS). This manifestation was part of a broader phenotype, which was recognized as a mild form of Beckwith-Wiedemann syndrome (BWS). Molecular analysis confirmed the diagnosis of BWS owing to a maternally inherited deletion of the centromeric imprinting center, or ICR2, an extremely rare genetic mechanism in BWS. The deletion interval (198 kb) also included exons 11–16 of the KCNQ1 gene, known to be responsible for LQTS at locus LQT1. No concomitant mutations were found in any other of the known LQT genes. The proposita’s mother carries the same deletion in her paternal chromosome and shows manifestations of the Silver-Russell syndrome (SRS). This report describes the smallest BWS-causing ICR2 deletion and provides the first evidence that a paternal deletion of ICR2 leads to a SRS-like phenotype. In addition, our observation strongly suggests that in cases of LQTS due to mutation of the KCNQ1 gene (LQT1), an accurate clinical genetic evaluation should be done in order to program the most appropriate genetic tests.
Similar content being viewed by others

Introduction
Sporadic and inherited long-QT syndromes (LQTS) are important causes of sudden cardiac death. In both cases, there is an abnormally prolonged cardiac repolarization interval, producing a potentially dangerous substrate for ventricular tachycardia and syncope or sudden death. These events may be triggered by physical and emotional stress.
Hereditary LQTS is an autosomal dominant disorder of the cardiac rhythm. Mutations in LQT genes occur in ∼1 in 2000 persons; however, as most mutation carriers remain asymptomatic throughout life, clinical disease is less frequent, affecting 1:3000–1:5000 individuals.1 The symptoms may present suddenly with syncope, seizures or even death as first manifestation.
Hundreds of mutations in >10 different genes have been associated with hereditary LQTS.2 Mutations in three of these genes, resulting in genetic subtypes designated LQT1, LQT2 and LQT3, respectively, account for the vast majority of cases.3 It has been suggested that the genotype, ie, the involved gene, and the site of mutation correlates with the clinical presentation.2
LQT1, the most common form, is caused by loss-of-function, heterozygous mutations in KCNQ1, which encodes a protein with structural features of a voltage-gated potassium channel.4 In LQT1 syndrome, syncope or sudden death is triggered by emotional or physical stress such as diving and swimming. When both KCNQ1 alleles are mutated, as in the Jervell and Lange-Nielsen syndrome (OMIM 220400), a severe prolongation of the QT interval occurs, increasing the risk of sudden death, and also causing congenital deafness.
The locus of KCNQ1 falls within a region of chromosome 11p subjected to imprinting and also involved in another genetic condition, the Beckwith-Wiedemann syndrome (BWS).5 The molecular bases of BWS are complex, and involve the altered expression of multiple growth regulatory genes in the 11p15.5 genomic region. Two imprinting domains (imprinting center 1 and 2, also referred to as ICR1 and ICR2) are responsible for regulating the expression of genes falling within this region. Interestingly, an antisense coding region, designated KCNQ10T1, exclusively expressed from the paternal allele, is located within the KCNQ1 locus, between exons 10 and 11. It corresponds to ICR2 and its RNA transcript blocks the expression of KCNQ1 and of the flanking gene CDKN1C, resulting in monoallelic expression of both genes from the maternal chromosome (Figure 1a). Imprinting defects at ICR2 (ie, hypomethylation of maternal KCNQ10T1) are responsible for about 50% of BWS cases.5

ICR2 regulation in normal and deleted individuals. Blue, paternal chromosome; Red, maternal chromosome. Small ovals correspond to putative enhancers for CDKN1C, one proximal and one remote. The position of the remote enhancer is inferred from the deletion interval in the family and differs from the previous report by Algar (2011). Green ovals indicate DMR (differentially methylated region), which can be methylated (black arch) or demethylated, to ligate an insulator (CTCF). (a) normally only the paternal antisense transcript KCNQ10T1,is expressed (arrow), whereas the maternal copy is silenced (X); paternal KCNQ10T1, acts as a repressor of the flanking CDKN1C growth suppressor gene and of the paternal KCNQ1 allele. As a consequence, only the maternal alleles of CDKN1C and KCNQ1 are expressed. The maternal transcription of CDKN1C is additionally regulated by at least 2 enhancers, one at a nearby position, the other one being putatively positioned more distantly. The precise location of these enhancers is yet unknown and is just proposed by Algar et al. Our deletion interval only overlaps with the one by Algar et al. for exons 10–15 of KCNQ1: as the strong enhancer is proposed within intron 3, our deletion interval should not include it. Alternatively, we propose that this remote enhancer might be located within the minimal deletion interval, ie, between exons 10 and 15. In the heart, the expression of KCNQ1 is expected to be biallelic because of tissue-specific imprinting regulation of this gene. (b) The proposita has a deletion including the maternal copy of KCNQ10T1, and part of the KCNQ1 maternal allele. The deletion interval likely includes the distal enhancer and, in addition, a repression effect in-trans of the paternal KCNQ10T1 cannot be ruled out. As a consequence, both CDKN1C alleles are downregulated (X), except for a mild residual enhancing effect excerpted by its nearby enhancer (dotted arrow). The paternal KCNQ1 allele is normally inactive and the maternal allele is inactivated by the deletion. Both alleles are thus inactive in most tissues. In the heart, because of this deletion, only the paternal allele of KCNQ1 is expected to be expressed in the heart leading to haploinsufficiency of this gene. (c) The proposita’s mother has a deletion including the paternal copy of KCNQ10T1, and part of the KCNQ1 paternal allele. As a consequence, given that the maternal KCNQ10T1, is normally inactive (X), both CDKN1C alleles are expressed (arrow) and the KCNQ1 maternal allele is normally active (arrow) in most tissues. In the heart, only the maternal allele of KCNQ1 is expected to be expressed leading to loss of function of this gene.
We report on a young woman admitted to our Cardiology Unit because of an episode of cardiac arrest related to LQTS. This manifestation was part of a broader phenotype, which was recognized as a mild form of BWS.
Materials and methods
Clinical report. A 20-year-old woman underwent a diagnostic cardiologic work-up after recurrent episodes of polymorphic, sustained ventricular tachycardia (VT) with ‘torsades de pointes’ morphology (Figure 2a), treated with multiple cardioverter-defribrillator shocks. Seven months before, she had been submitted to a cardiological evaluation because of episodes of chest pain, and was found to have hypertension. The EKG showed the presence of depolarization abnormalities (ie, negative T waves) in lateral leads (Figure 2b), whereas a 2D-echocardiogram was substantially normal and laboratory tests were unremarkable. She was discharged on antihypertensive therapy with carvedilol and amlodipine. Nevertheless, 2 months later, she experienced new VT episodes and a syncope attack with palpitations at its onset.

(a) Polymorphic ventricular tachycardia (torsades de pointes) occurred during monitoring. (b) ECG record showing a long Q-T interval (arrow). (c) Patient’s facial appearance. Note that the face appears coarse, with prominent mandible and M-shaped upper lip.
On re-admission to our Cardiology Unit, the clinical examination was substantially normal and the EKG showed the presence of sinus rhythm with depolarization abnormalities in lateral leads with marked QT prolongation (QT 470 ms with HR 73 bpm, QTc 495 ms). The patient denied the assumption of any drugs known to prolong the QT interval and laboratory tests failed to detect the presence of electrolyte abnormalities. A 2D-echocardiogram was normal and cardiac magnetic resonance with contrast-enhanced sequences confirmed the absence of structural heart anomalies. In addition, the patient underwent a complete screening for the presence of secondary causes of hypertension that failed to detect any relevant findings. On the basis of medical history, instrumental and laboratory findings, we ruled out the presence of acquired causes of QT prolongation and, therefore, a diagnosis of genetic LQTS was proposed. Before discharge, the patient underwent dual-chamber intracardiac defibrillator implantation for secondary prevention and medical therapy with metoprolol was started.
First-degree relatives screening allowed the detection of a mild QT prolongation (QT 445 ms with HR 69 bpm, QTc 463 ms) with flat T waves in V2–V3 in the patient’s mother, a 56-year-old woman, who had no history of syncope attacks or palpitation and whose Holter monitoring did not reveal any significant arrhythmia.
The unusual physical phenotype of the patient (supplementary data) prompted the cardiologists to request a clinical genetic evaluation, which demonstrated the following: height 178 cm (>97th centile), weight 46 kg (<3rd centile), head circumference 51.5 cm (<3rd centile). The maternal height was 152 cm (<3rd centile) and the paternal height was 177 cm (50th–75th centile). The patient’s face appeared ‘coarse’ with proptosis, apparent hypotelorism and mild upslanting of the palpebral fissures, ‘M’ shape of the upper lip, wide mandible, with apparent increase above normal of the inter-gonial diameter and mild prognathism. Ears were posteriorly angulated and there were creases in the anterior face of the ear lobe (Figure 2c).
All these findings led us to hypothesize that the underlying genetic defect for the LQTS was a dysfunction of the LQT1 gene in the BWS region.
MLPA and high resolution Array-CGH analysis. To confirm the tentative diagnosis of BWS, methylation-sensitive MLPA analysis was performed with a set of probes specific for the BWS critical region.
Methylation-sensitive MLPA analysis was performed with a commercially available kit specific for the diagnosis of Beckwith Wiedemann/Silver-Russell syndromes (SALSA MS-MLPA ME030-B2 BWS/RSS).6 Conditions for testing and statistical analysis were as recommended by the manufacturer. The test was also performed in the patient’s parents.
High-resolution (35 kb) array-CGH with the 244K-kit from Agilent was performed in order to refine the size of the deletion that had been detected by MLPA. Conditions for testing and statistical analysis were as recommended by the manufacturer.
Sanger-sequencing was performed by standard methods on KCNQ1 and on eight additional LQT genes: HERG, MINK, MIRP1, MIRP2,SCN4B, KCNJ2, SNTA1, CAV3.
Results
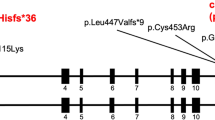
A microdeletion at the ICR2 region was detected by MLPA. The size of the deletion was 198 kb, encompassing the entire KCNQ1OT1 antisense (AS) transcript, as well as exons 11–16 of KCNQ1. Interestingly, the ICR2 deletion was inherited from the mother, who carries the same alteration on the paternal allele, as established by the methylation pattern of the residual allele (Figure 3a and b). High-resolution array-CGH confirmed the results of MLPA analysis, defining a 198-Kb deletion from probe A_16_P02381662 to probe A_16_P02382007. No other genes were comprised in the deletion interval (Figure3c). No pathogenic alterations were detected by sequencing the other LQT genes, except for a number of polymorphisms (Supplementary table 1).

MLPA and array-CGH results. MLPA histogram shows the probes deleted in the proposita (a) and in her mother (b). These probes correspond to the KCNQ10T1,locus and to three exons (12, 15, 16) of the KCNQ1 gene in the BWS-RSS region. (c) High-resolution array-CGH plot. Dark green rectangle indicates the ADM-2 algorithm call of the 11p15.5 deletion, of about 198 Kb, from 2 671 855–2 870 191 genomic position (NCBI 37, UCSC hg19). This deletion encompasses the KCNQ1 gene from intron 11 to exon 16, and includes entirely the region encoding the KCNQ1OT1 antisense RNA.
Discussion
BWS is an overgrowth syndrome, usually presenting neonatally with omphalocele, macroglossia and macrosomia, but frequently characterized by milder clinical signs that may not be readily recognized, especially in adulthood. An increased risk for childhood tumors has to be considered when this diagnosis is established. The molecular etiology of BWS is quite complex: about 80% of cases are due to genomic and/or epigenomic alterations in one of the two imprinting centers, ICR1 and ICR2, in the distal short arm of chromosome 11.5 In the present patient, an intragenic deletion causing loss of function of KCNQ1 is likely responsible for LQTS, whereas the complete ablation of the KCNQ1OT1 antisense transcript on the maternal chromosome likely causes the physical anomalies typical of BWS. Deletions of ICR2 are extremely rare causes of this condition, whereas an imprinting defect (ie, an hypomethylation of maternal ICR2) is found in about 50% of patients with this diagnosis.5
This is, to our knowledge, the third report of an inherited deletion specifically and exclusively involving ICR2. The first one7 was found in a family, where a carrier mother had three children affected with BWS due to downregulation of the maternal copy of CDKN1C, consequent to the deletion of ICR2. A similar ICR2 deletion was also reported in a second family in which maternal transmission led to a BWS phenotype in the offspring because of underexpression of CDKN1C.8 This gene is commonly expressed by the maternal chromosome in which ICR2 is methylated, thus preventing chromatin insulator formation and allowing a distant enhancer cis-activity on CDKN1C transcription. It has been speculated that the maternal ICR2 deletion either exposes CDKN1C to repression in trans by the paternal KCNQ10T1 antisense, or removes a cis-acting distant enhancer of CDKN1C expression. This postulated enhancer has been tentatively located within intron 3 of the KCNQ1 gene.8 Both pathogenic mechanisms are likely to occur in our BWS patient: we speculate that absence of the maternal KCNQ1 transcript does not protect the flanking CDKN1C from the silencing effect of the paternal antisense transcript. According to this hypothesis, a cis-protection effect preventing downregulation might be exerted by ICR2 on flanking genes in the maternal chromosome. Similarly, the deletion of the maternal CDKN1C distant enhancer may also be responsible for the lack of maternal expression of this gene. A more proximal enhancer with milder effect on CDKN1C transcription is predicted at 3′ of KCNQ1. If the position in intron 3 of the distant enhancer is correct, then this enhancer should not be deleted in our proposita; however, we propose that the deletion interval in our patient includes this strong enhancer thus suggesting a different position, not in intron 3, as previously proposed,8 but rather to a more restricted interval, between intron 10 and 15. The loss of a strong enhancer and the maintenance of a weak one might explain the mild expression of the BWS phenotype related to some residual expression of CDKN1C. The possible mechanism of alteration of gene expression in this imprinted region is shown in Figure 1b and c), both in the proposita and her mother. According to this model, the proposita should have biallelic repression of both the CDKN1C and the KCNQ1 gene in all tissues except the heart, where only the paternal copy of KCNQ1 should be expressed, in accordance with mouse data, indicating that this gene is not imprinted in the cardiac muscle.9 The proposita’s mother should have biallelic expression of the CDKN1C gene and monoallelic expression of the maternal KCNQ1 allele in all tissues, including the heart.9
In the family reported by Niemitz et al.7 the sibs with BWS did not have a cardiac phenotype and their mother, who inherited the paternal ICR2 deletion, did not have any phenotypic anomaly. The same phenotypic manifestations were observed in the family reported by Algar et al.8 (2011). On the contrary, in our family the proposita had severe LQTS and mild BWS, whereas, the carrier mother had short stature and relative macrocephaly, in addition to LQT interval, which had never been symptomatic, at least until the time of our observation. This suggests that the absence of the paternal KCNQ1 antisense leads to upregulation of the CDKN1C in cis: therefore, the mother of the proposita is likely to have biallelic expression of this gene, and consequently excessive growth repression, leading to a SRS-like phenotype. Accordingly, it has been shown in the mouse that a targeted deletion of the paternal Kcnq10t1 allele leads to the overexpression of the normally silent Cdkn1c paternal allele.10 The observation that an isolated ICR2 alteration leads to a SRS-like phenotype strenghtens the debated hypothesis that ICR2 itself contributes to SRS manifestations through upregulation of CDKN1C and possibly other genes within the same cluster. Gain of function of CDKN1C seems to be a major cause of restricted growth, as also suggested by the recent report of gain-of-function mutations in patients with IMAGe syndrome, a condition strikingly similar to SRS, characterized by growth retardation, skeletal dysplasia, adrenal hypoplasia and genital anomalies.11,12
Because of the severity of LQT clinical presentation in the proposita with respect to the asymptomatic LQT in her mother, we speculated that this might be due to additional mutations either in the paternally inherited KCNQ1 allele or in other genes known to cause LQT. The sequence analysis of these genes did not detect pathogenic mutations, thus disclaiming this hypothesis. However, the observed difference in clinical severity might still be due to mutations in other unknown LQT genes or, alternatively, to an imprinting disregulation occurring in the heart. As already mentioned, while it is generally held that KCNQ1 has biallelic expression in the heart muscle, this has been demonstrated only in the mouse.9 If that were not the case in man, with monoallelic expression of KCNQ1 in the heart muscle as in all other tissues, then the proposita would have no KCNQ1 functioning allele in the heart, explaining the severity of her cardiac condition, whereas her mother would still have monoallelic expression in the heart from the maternal chromosome (the deletion being in the normally silent paternal allele), thus explaining the less severe cardiac phenotype.
Given the biallelic inactivation of KCNQ1 in the proposita, one should expect phenotypic manifestations of the Jervell and Lange-Nielsen syndrome, an autosomal recessive disorder characterized by congenital deafness, LQT and high risk for cardiac arrhythmias. Our proposita had normal hearing, likely because the paternal KCNQ1 allele is normally expressed.
We reported another case of a maternally inherited ICR2 deletion13 that included CDKN1C, as well as additional genes, clearly explaining the BWS phenotype because of lack of the maternal CDKN1C transcript. This patient was recently recalled to measure the QT interval, that was found to be normal.
As practical conclusion, our proposita was counseled a 50% recurrence risk of BWS and LQTS in her offspring. In addition, we suggest that in cases of LQTS an accurate clinical genetic evaluation should be done in order to program the most appropriate genetic tests and follow up. In this specific case, molecular analysis of the LQT1 gene by standard sequencing would have given normal results because only the residual allele of LQT1 would have been sequenced.
References
Roden DM : Human genomics and its impact on arrhythmias. Trends Cardiovasc Med 2004; 14: 112–116.
Moss AJ, Kass RS : Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest 2005; 115: 2018–2024.
Roden DM : Clinical practice. Long-QT syndrome. N Engl J Med 2008; 358: 169–176.
Wang Q, Curran ME, Splawski I et al: Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996; 12: 17–23.
Choufani S, Shuman C, Weksberg R : Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010; 154C: 343–354.
Scott RH, Douglas J, Baskcomb L et al: Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with overgrowth and growth retardation. J Med Genet 2008; 45: 106–113.
Niemitz EL, DeBaun MR, Fallon J et al: Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am J Hum Genet 2004; 75: 844–849.
Algar E, Dagar V, Sebaj M et al: An 11p15 imprinting centre region 2 deletion in a family with Beckwith Wiedemann syndrome provides insights into imprinting control at CDKN1C. PLoS One 2011; 6: e29034.
Lee MP, Hu RJ, Johnson LA et al: Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat Genet 1997; 15: 181–185.
Horike S, Mitsuya K, Meguro M et al: Targeted disruption of the human LIT1 locus defines a putative imprinting control element playing an essential role in Beckwith-Wiedemann syndrome. Hum Mol Genet 2000; 9: 2075–2083.
Arboleda VA, Lee H, Parnaik R et al: Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet 2012; 44: 788–792.
Riccio A, Cubellis MV : Gain of function in CDKN1C. Nat Genet 2012; 44: 737–738.
Zollino M, Orteschi D, Marangi G et al: A case of Beckwith-Wiedemann syndrome caused by a cryptic 11p15 deletion encompassing the centromeric imprinted domain of the BWS locus. J Med Genet 2010; 47: 429–432.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Gurrieri, F., Zollino, M., Oliva, A. et al. Mild Beckwith-Wiedemann and severe long-QT syndrome due to deletion of the imprinting center 2 on chromosome 11p. Eur J Hum Genet 21, 965–969 (2013). https://doi.org/10.1038/ejhg.2012.280
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.280
Keywords
This article is cited by
-
Unusual deletion of the maternal 11p15 allele in Beckwith–Wiedemann syndrome with an impact on both imprinting domains
Clinical Epigenetics (2021)
-
Overlap phenotypes of the left ventricular noncompaction and hypertrophic cardiomyopathy with complex arrhythmias and heart failure induced by the novel truncated DSC2 mutation
Orphanet Journal of Rare Diseases (2021)
-
A paternally inherited 1.4 kb deletion of the 11p15.5 imprinting center 2 is associated with a mild familial Silver–Russell syndrome phenotype
European Journal of Human Genetics (2021)
-
Glucocorticoid receptor triggers a reversible drug-tolerant dormancy state with acquired therapeutic vulnerabilities in lung cancer
Nature Communications (2021)
-
Frequency of KCNQ1 variants causing loss of methylation of Imprinting Centre 2 in Beckwith-Wiedemann syndrome
Clinical Epigenetics (2020)
