
Abstract
Thoracic aortic aneurysms and dissections (TAAD) is a serious condition with high morbidity and mortality. It is estimated that 20% of non-syndromic TAAD cases are inherited in an autosomal-dominant pattern with variable expression and reduced penetrance. Mutations in myosin heavy chain 11 (MYH11), one of several identified TAAD genes, were shown to simultaneously cause TAAD and patent ductus arteriosus (PDA). We identified two large Dutch families with TAAD/PDA and detected two different novel heterozygote MYH11 variants in the probands. These variants, a heterozygote missense variant and a heterozygote in-frame deletion, were predicted to have damaging effects on protein structure and function. However, these novel alterations did not segregate with the TAAD/PDA in 3 out of 11 cases in family TAAD01 and in 2 out of 6 cases of family TAAD02. No mutation was detected in other known TAAD genes. Thus, it is expected that within these families other genetic factors contribute to the disease either by themselves or by interacting with the MYH11 variants. Such an oligogenic model for TAAD would explain the variable onset and progression of the disorder and its reduced penetrance in general. We conclude that in familial TAAD/PDA with an MYH11 variant in the index case caution should be exercised upon counseling family members. Specialized surveillance should still be offered to the non-carriers to prevent catastrophic aortic dissections or ruptures. Furthermore, our study underscores that segregation analysis remains very important in clinical genetics. Prediction programs and mutation evaluation algorithms need to be interpreted with caution.
Similar content being viewed by others


Introduction
Thoracic aortic aneurysms and dissections (TAAD) is a serious condition with high morbidity and mortality.1, 2 Aortic medial degeneration, histopathologically characterized by loss of elastin fibers and smooth muscle cells and increased proteoglycan deposition, is thought to precede TAAD.3 TAAD can be classified as sporadic, syndromic and familial non-syndromic.4 Sporadic forms of TAAD tend to develop at an older age and their main risk factor is hypertension.5 The syndromic forms of TAAD include the connective tissue diseases Marfan syndrome, Loeys-Dietz syndrome, the vascular type of Ehlers-Danlos syndrome, and the newly described aneurysms osteoarthritis syndrome (AOS), which are caused by mutations in the FBN1, TGFBR1/TGFBR2, COL3A1 and SMAD3 genes, respectively.4, 6, 7
In the non-syndromic TAAD cases, 20% is estimated to be familial with an autosomal-dominant inheritance pattern (FTAAD).8 FTAAD is a heterogeneous and complex condition with variable expression and reduced penetrance.9 Up until now, seven loci have been linked to familial TAAD and from these loci four genes have been identified; TGFBR1, TGFBR2, MYH11 (myosin heavy chain 11) and ACTA2.4, 7 Recently, two other TAAD genes have been identified. MYLK was discovered by a candidate gene sequencing approach.10 Mutations in SMAD3 were first identified by traditional linkage analysis followed by candidate gene sequencing in patients with syndromic TAAD.7 Later on SMAD3 mutations were also discovered by whole-exome sequencing in familial non-syndromic TAAD.11 All six genes can be analyzed in diagnostic settings when familial TAAD is suspected. Identification of a causal mutation in the index case will have great implications. First, it will enable the genetic screening of family members, which will allow non-carriers to be excluded from further clinical monitoring. Second, for mutation carriers, the identification of a causal mutation will influence pharmaceutical treatment and decision-making concerning surgical treatment.12
MYH11 and ACTA2 encode for specific vascular smooth muscle cell (VSMC) components. In aortic tissue, in which these genes were disrupted, VSMC loss, disorganization and hyperplasia were observed.13, 14, 15, 16 Additionally, carriers of MYH11 and ACTA2 mutations may present with a patent ductus arteriosus (PDA),13, 14 possibly as a result of an impaired ability of the VSMC to proliferate, migrate and/or contract.17 MYH11 mutations are thought to account for only 1% of the familial TAAD cases and until now they have only been described in a few families, which had both TAAD and PDA cases and not in families with TAAD as the only vascular phenotype.15
Here, we describe two large TAAD/PDA families in which MYH11 variants do not fully segregate with the vascular phenotype and discuss its clinical implications.
Materials and methods
Patients
Parents of the probands were referred to our Department of Clinical Genetics by their specialist. Initial pedigrees were constructed. Patients were examined for associated skeletal and skin features of connective tissue disorders. First- and second-degree family members of persons with TAAD/PDA were referred to a cardiologist for monitoring according to standard recommendations.12 Upon consent, all relevant medical information was retrieved. The diagnosis of TAAD/PDA was determined by post mortem section, ultrasound, CT or MRI imaging and the aortic diameters were measured in the sinus of Valsalva or in the ascending aorta (Table 1). Individuals were marked as affected if they had a ruptured thoracic aortic aneurysm, a dissection type A or B and/or a PDA. As no complete information on body surface area was available, individuals in whom the sinus of Valsalva or the ascending aorta had a diameter >42 mm were marked as affected. Persons with mild dilatations, of whom there could be doubt about affected status, are not expected to be classified as affected with this wide cutoff.18 Individuals at risk for inheriting the condition with normal or unknown aortic diameters were considered as ‘unknown’ because of the variable expression and reduced penetrance of the condition. After obtaining informed consent, blood was collected and genomic DNA was isolated according to standard procedures.
Genetic testing and segregation analysis
Candidate TAAD genes were sequenced in the proband of family TAAD01 and/or his affected brother, in the mother of the proband of family TAAD02 and in individuals without the MYH11 mutations in a diagnostic setting (VUMC, The Netherlands). MYH11 mutations were subsequently sequenced in all available family members (TAAD01: 24 individuals, TAAD02: 16 individuals) as well as in control chromosomes by standard Sanger sequencing. Primer information is available upon request.
As rare copy nuber variants have recently been associated with susceptibility to TAAD development,19, 20 array-CGH (comparative genome hybridization) analysis was performed in individual VI:1 of family TAAD01 and individual III:4 of family TAAD02 using 105K microarray slides from Agilent Technologies (Santa Clara, CA, USA) following manufacturer’s protocols. Data analysis was performed using DNA analytics 4.76 software from Agilent technologies using the ADM-2 algorithm. Interpretation of CNV-data was performed as described in the guidelines of Vermeesch et al.21 The resolution was ∼50 kb.
The presence of intragenic deletions or duplications in SMAD3 in individual III:4 of family TAAD02 was tested by QPCR on a lightcycler LC480 384 well (Roche, Basel, Switzerland), using primers that amplify single exons of SMAD3 and FBN1 (sequences available on request). Melting curve analysis was used to confirm presence of a single PCR product in each well. The quantification of SMAD3 exons was compared with exons of FBN1 in which deletions/duplications had been excluded by MLPA testing. A standard curve for quantification was created by amplification of dilutions of a standardized DNA solution of 20 ng/μl with PCR fragments of FBN1 and SMAD3. The QPCR was validated by confirming deletions in FBN1 or SMAD3 that had been found in other patients by MLPA or microarray.
Genome-wide genotyping of SNP markers and non-parametric linkage analysis
DNA of twenty-four family members of family TAAD01 was genotyped on Illumina’s SNP-based linkage panel V (Illumina Inc., San Diego, CA, USA), including 6056 informative SNP markers distributed evenly across the human genome with a median genetic distance of 0.38 cM and a physical distance of 341 kb, using the BeadArray technology on an Illumina BeadStation following the manufacturer’s protocol (www.illumina.com). DNA of 16 family members of family TAAD02 was genotyped on Illumina’s HumanCytoSNP-12v2_A Genotyping BeadChip SNP array and analyses were performed according to the protocol of the manufacturer (Illumina Inc.). All SNPs were examined for their genotyping quality and those that had a low signal or were poorly clustered or were not polymorphic were excluded before analysis.
Data for family TAAD02 were pruned to 13 156 autosomal polymorphic markers with median distance of 0.13 cM or 116 kb.
Non-parametric multipoint linkage (NPL) analysis was carried out with Merlin (v.1.1.2)22 on a reduced pedigree to accommodate limitations of the software and DNA availability (Figure 1 and Table 1). Related individuals with normal or unknown aortic diameters were marked as ‘unknown’ phenotypes. Spouses were marked as ‘unaffected’. Results report NPL LOD-scores. Linkage results at candidate genes/loci are reported as the highest NPL LOD-score (interpolated) within the boundaries of the gene/locus.

Pedigrees of family TAAD01 (a) and family TAAD02 (b) that participated in the study demonstrating an autosomal-dominant inheritance with reduced penetrance. Affected family members in whom the MYH11 variant was not present are indicated with a light blue arrow, red asterisks indicate individuals used in NPL analysis.
Results
Clinical evaluations
Family TAAD01
Family TAAD01 was a seven-generation Dutch family, in which TAAD had been diagnosed in two generations (V and VI) and a PDA in one generation (VII) (Figure 1a and Table 1). Sudden death occurred frequently in generation IV between the ages of 40 and 70 years, but no autopsies were performed to explore the causes of these deaths. The proband (VI:2) died of complications of an emergency TAA repair at the age of 25. Subsequently, his brother (VI:1) was diagnosed with a TAA >80 mm and a dissection type A at the age of 24, which was successfully repaired. The parents of the proband were related with a kinship coefficient of 1/128. In the family of father (V:2) no aortic disease was documented. In the family of mother (V:3) there were several additional confirmed cases of TAAD and one case of PDA. Her brother (V:5) died from an acute dissection type A at 28 years of age. Her cousin (V:9) underwent TAAD repair at the age of 70 and his brother (V:8) died of a dissection type A at the age of 70. Their sister (V:11) had emergency repair for a type A dissection at the age of 70. In another cousin of the mother of the proband (V:20) a chronic dissection type B was diagnosed at the age of 58, which was conservatively treated. He died at the age of 63, possibly of a cerebrovascular accident. No autopsy was performed. His sister (V:19) underwent surgery for an acute dissection type A at the age of 59. Another sister (V:18) died from a ruptured TAA at the age of 52. Their nephew (VI:8) died of a dissection type B at the age of 47. His daughter (VII:1) was recently diagnosed with a PDA at the age of 35.
By cardiological assessment of first- and second-degree family members of TAAD patients, thoracic aortic aneurysms were discovered in the mother of the proband V:3, (44 mm), her brother V:6 (44 mm) and another cousin V:15 (47 mm). The latter had no history of hypertension and recently underwent prophylactic surgical repair of his TAA (Bentall procedure). None of the clinically evaluated family members exhibited skin or skeletal signs of a connective tissue disease.
Family TAAD02
Family TAAD02 was a five-generation Dutch family, in which both TAAD (III and IV) and PDA (III and V) had been diagnosed in two generations (Figure 1b and Table 1). Unfortunately, no medical data or information about causes of deaths of generations I and II could be retrieved. The proband (IV:3) died of an acute type A dissection at the age of 18. His mother (III:7) was diagnosed with a PDA at the age of 5. One of her three sisters (III:10) had a ductal diverticulum on a MRI, which we considered as an abortive form of PDA, and her only brother (III:11) also had a PDA. The daughter of the half-brother of the proband (V:1), who was born at term, was also diagnosed with a PDA and recently underwent a reparative surgery. Her father (IV:2) was diagnosed with a mild pulmonary stenosis. A cousin of the mother of the proband (III:4) had pathological dilated ascending aorta that was repaired when it exceeded 50 mm in diameter at the age of 50. Histological examination revealed extensive medial degradation with mucoid accumulation. This patient was not known to be hypertensive.
None of the clinically evaluated family members exhibited skin or skeletal signs of a connective tissue disease.
Sequencing and segregation analysis of TAAD candidate genes
Family TAAD01
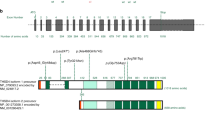
No mutation in the TGFBR1, TGFBR2, FBN1, COL3A1, ACTA2, MYLK and SMAD3 genes was discovered in the proband or his affected brother. In MYH11 a heterozygous missense variant was found: a nucleotide substitution 232A>G in an AAG triplet encoding the basic lysine leading to a GAG triplet encoding the acidic glutamic acid (c. 232A>G, p. K78E). The variant is localized in a highly conserved block, just one amino acid before the start of the ATP-binding motor domain of the myosin head (Figure 2a). The Genomic Evolutionary Rate Profiling (GERP) scores were 3.38 for the 19-way GERP score and 3.7 for the 34-way GERP score (www.ensembl.org), which indicates a high degree of revolutionary conservation across several species down to C. elegans. PolyPhen2 and SIFT analysis of this alteration predicted a probably damaging and damaging effect, respectively. Segregation analysis revealed that the MYH11 variant was present in most TAAD cases in this family (n=8). However, it was not identified in three affecteds: an individual with a TAA (V:15), an individual with a chronic dissection type B (V:20) and an individual with a PDA (VII:1). The 232A>G variant was not present in 200 control alleles and has not been reported in dbSNP131, the 1000 Genome Project (www.1000genomes.org) and our in-house whole-exome sequencing database, consisting of 60 whole-exomes of Dutch origin(MH, IJN, EC). The variant was also not present within the Exome Variant Server database (http://snp.gs.washington.edu/EVS).

Representation of the MYH11 variants identified in family TAAD01 (a) and TAAD02 (b), including protein domain localization and high evolutionary conservation.
In the three individuals without the MYH11 variant, TGFBR1, TGFBR2, ACTA2, MYLK and SMAD3 were sequenced. No mutation was identified.
Array-CGH analysis in individual VI:1 showed no rare deletion or duplication.
Family TAAD02
No mutation was detected in the ACTA2 gene in the mother of the proband (III:7). However, in MYH11 a heterozygous deletion of an AAG triplet from a conserved stretch of four lysines 1256K localizing in the prefoldin domain (part of the chaperone system) of myosin tail was discovered (Figure 2b). Conservation scores for the three deleted nucleotides are 1.45, 2.59 and 2.59 for the 19-way GERP score and 0.98, 3.45 and 2.36 for the 34-way GERP score indicate a high evolutionary conservation. PolyPhen2 and SIFT analysis of this alteration is not possible as these programs do not predict the effect of (in-frame) deletions. Segregation analysis revealed that the MYH11 variant was also present in the proband (IV:3) and in two cases of PDA (III:10 and III:11). However, it was not identified in the individual with the pathological aortic dilatation (III:4) and neither in another PDA case (V:1). The 3766-68delAAG variant was not present in 800 control alleles and has not been reported in the 1000 Genome Project (www.1000genomes.org) and our in-house whole-exome sequencing database. Additionally, in case III:4 ACTA2, FBN1, TGFBR1, TGFBR2, MYLK and SMAD3 were analyzed, but no mutation was identified.
Array-CGH analysis in individual III:4 showed no rare deletion of duplication.
Non-parametric linkage analysis in TAAD candidate genes
Non-parametric LOD-scores (NPL) were checked for loci and genes that have previously been shown to be involved in TAAD. None of these loci showed NPL-scores near the maximum obtainable NPL of 1.84 for family TAAD01 (Table 2). As informativeness was very high, this suggested there was no complete sharing of these regions between all affected family members. Reconstruction of the most likely haplotypes in these regions supported the assumption that not all affected individuals shared the same haplotype in any of these regions (data not shown), including the MYH11 locus (Supplementary Figure). The NPL LOD-score of this locus was 1.0, which is consistent with partial sharing of a haplotype between affected family members. Segregation analysis of p. K78E had already revealed incomplete sharing of this variant between affected family members. In accordance, haplotype reconstruction of the MYH11 locus indeed indicated incomplete sharing of the full MYH11 gene between affected family members (Supplementary Figure). It is therefore not expected that another cryptic mutation in the MYH11 gene, shared by all TAAD patients within this family, is responsible for the NPL LOD-score of 1.0 in this region.
In family TAAD02, NPL analysis showed complete sharing at a 19-Mb locus on chromosome 15 (NPL Z=5.9, NPL LOD=0.9), encompassing the SMAD3 gene, but many other genes as well. Though sharing was estimated to be complete, supported by best guess haplotypes, this was not statistically significant, owing to the small family size. The SMAD3 gene was sequenced in individual III:4, but no mutation was identified. Array-CGH analysis in the same patient revealed no large deletion or duplication within this region. Similarly, intragenic deletion and duplication were excluded by QPCR.
Discussion
We have clinically characterized two independent large Dutch families with TAAD and PDA. In routine diagnostic testing a MYH11 variant was found in both families. However, segregation analysis revealed that some individuals with a vascular phenotype did not carry the variant.
TAAD is usually an asymptomatic condition until a life-threatening event with high mortality occurs (ie, aortic dissection or rupture). Therefore, genetic counseling and specialized monitoring of families with inherited forms of TAAD/PDA is crucial. Identification of a causal gene in the family will have implications for screening and pharmaceutical or surgical treatment.9 However, assigning a pathogenic potential to a given variant can be challenging. When functional assays are not readily available, segregation analysis in additional family members is usually the only way to claim causality of a variant predicted to be pathogenic by current mutation evaluation algorithms. However, families may not be large enough to solidly perform this type of analyses. In addition, the variable expression and reduced penetrance of TAAD/PDA complicates segregation patterns as mutation carriers may not (yet) have developed clinical phenotypes. Furthermore, even though standard values, stratified for gender and body surface area, of the diameter of different thoracic aortic segments are available there is still a gray area in which a person with a mildly dilated aorta can be considered as affected or not.
Circumstantial evidence for pathogenicity of a mutation can be obtained from conservation scores and bioinformatic predictions of the effect of the variant on protein structure. The variants discovered in our study were either resulting in substitution or loss of a lysine in highly conserved regions, with the missense variant of family TAAD01 affecting the head and the in-frame deletion of family TAAD02 the tail of myosin. The families reported here were thoroughly clinically examined and large enough to conclude that the identified MYH11 variants could not fully explain the vascular phenotypes observed. However, in general practice substantial clinical information and multiple (affected) family members may not be available and therefore incorrect conclusions about causality of variants may be drawn. As an example, we experienced an unexpected diagnostic turn in family TAAD02 when vascular disease in the two non-carriers III:4 and V:1 was discovered after the MYH11 variant originally was present in all affected family members that were used for segregation analysis in the early stage. Luckily, an aortic dilatation was detected in individual III:4 and prophylactic surgery was performed before she was released from cardiological follow-up.
There are three possible explanations for our findings. First, the MYH11 variants are not pathogenic and do not contribute to the initiation of the disease in the carriers at all. If this is the case, taking into account the mode of inheritance in the pedigrees, there is most likely another monogenic factor that is responsible for the TAAD/PDA. However, because of the pathogenic prediction of the alterations by mutation evaluation programs and/or conservation scores, it is tempting to assume that there is an effect of the alterations. Moreover, it would be very coincidental to find novel alterations in MYH11 within these TAAD/PDA families that do not contribute to the vascular phenotype at all. A second explanation of our findings is that the MYH11 variants are fully responsible for the vascular disease in the carriers and that in affected non-carriers in the described families the phenotype is caused by other genetic or environmental factors. However, because the incidence of TAAD is low, especially at an age below 60 years, as is the prevalence of PDA in the adult population, this is not a very plausible explanation.5, 23 Furthermore, mutations in the known TAAD genes were excluded in the non-carriers by mutation analysis.
A third possibility is that the MYH11 variants are responsible for part of the phenotype and that there are additional genetic factors that can lead to the disease either by themselves or by interaction with the MYH11 mutation, so operating in a di- or even multigenic model. The theory of an oligogenic disease would also explain the generally variable onset and severity as well as the reduced penetrance between and within TAAD families.7, 8 We therefore consider this third explanation as the most likely one. Further research of the described families including whole- exome sequencing may reveal the explicit genetic causes of TAAD/PDA. In family TAAD02, NPL analysis showed complete sharing of a region of 19 Mb on chromosome 15 encompassing the SMAD3 gene. Subsequent mutation analysis and QPCR of SMAD3 did not reveal a mutation or an intragenic deletion/duplication, suggesting that SMAD3 is probably not the gene causing TAAD within this family. Other genes within this 19 Mb region may contribute to the vascular phenotype, but the power is too small to reach significant NPL results because of the limited number of affected family members.
In summary, we present two examples of large families with incomplete segregation of independent MYH11 variants with TAAD/PDA. We hypothesize that within these families additional genetic factors are involved in the etiology of the disease, acting as an oligogenic model. Such a model would also explain the variability of the onset and progression of the disorder and its reduced penetrance in general. Recently upcoming high-throughput sequencing technologies such as (whole) exome or even whole-genome sequencing may help to uncover the genetic basis in the families described in this report, as well as in other TAAD families that have remained unresolved. For now, our main message is that caution should be exercised when family members who do not carry a family-specific MYH11 variant are counseled, as they cannot be reassured and should still participate in specialized surveillance programs. Furthermore, our study underscores that segregation analysis remains very important in clinical genetics. Prediction programs and mutation evaluation algorithms should be interpreted with caution.
References
Lilienfeld DE, Gunderson PD, Sprafka JM, Vargas C : Epidemiology of aortic aneurysms: I. Mortality trends in the United States, 1951 to 1981. Arteriosclerosis 1987; 7: 637–643.
Lindsay J : Diagnosis and treatment of diseases of the aorta. Curr Probl Cardiol 1997; 22: 485–542.
Guo DC, Papke CL, He R, Milewicz DM : Pathogenesis of thoracic and abdominal aortic aneurysms. Ann N Y Acad Sci 2006; 1085: 339–352.
El-Hamamsy I, Yacoub MH : Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol 2009; 6: 771–786.
Ince H, Nienaber CA : Etiology, pathogenesis and management of thoracic aortic aneurysm. Nat Clin Pract Cardiovasc Med 2007; 4: 418–427.
Lindsay ME, Dietz HC : Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011; 473: 308–316.
van de Laar IM, Oldenburg RA, Pals G et al: Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011; 43: 121–126.
Albornoz G, Coady MA, Roberts M et al: Familial thoracic aortic aneurysms and dissections – incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82: 1400–1405.
Milewicz DM, Chen H, Park ES et al: Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am J Cardiol 1998; 82: 474–479.
Wang L, Guo DC, Cao J et al: Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 2010; 87: 701–707.
Regalado ES, Guo DC, Villamizar C et al: Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res 2011; 109: 680–686.
Hiratzka LF, Bakris GL, Beckman JA et al: 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons,and Society for Vascular Medicine. J Am Coll Cardiol 2010; 55: e27–e129.
Zhu L, Vranckx R, Khau Van Kien P et al: Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 2006; 38: 343–349.
Guo DC, Pannu H, Tran-Fadulu V et al: Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007; 39: 1488–1493.
Pannu H, Tran-Fadulu V, Papke CL et al: MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet 2007; 16: 2453–2462.
Milewicz DM, Guo DC, Tran-Fadulu V et al: Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet 2008; 9: 283–302.
Slomp J, Gittenberger-de Groot AC, Glukhova MA et al: Differentiation, dedifferentiation, and apoptosis of smooth muscle cells during the development of the human ductus arteriosus. Arterioscler Thromb Vasc Biol 1997; 17: 1003–1009.
Sheil ML, Jenkins O, Sholler GF : Echocardiographic assessment of aortic root dimensions in normal children based on measurement of a new ratio of aortic size independent of growth. Am J Cardiol 1995; 75: 711–715.
Prakash SK, LeMaire SA, Guo DC et al: Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet 2010; 87: 743–756.
Kuang SQ, Guo DC, Prakash SK et al: Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet 2011; 7: e1002118.
Vermeesch JR, Fiegler H, de Leeuw N et al: Guidelines for molecular karyotyping in constitutional genetic diagnosis. Eur J Hum Genet 2007; 15: 1105–1114.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin – rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Ng AS, Vlietstra RE, Danielson GK, Smith HC, Puga FJ : Patent dusctus arteriosus in patients more than 50 years old. Int J Cardiol 1986; 11: 277–285.
Acknowledgements
We are grateful to the patients and their relatives who participated in our study. We thank Drs E van Binsbergen for array-CGH analysis. Our research was supported by a grant from the foundation Nuts Ohra (SNO-T-0701-101). AF Baas was supported by a grant from the Dr E Dekker Program of the Netherlands Heart Foundation (2009T001). X Jeunemaitre is supported by grants from ANR (n°10-02, GDPM2) and from the European FP7 through the FAD integrated project (Fighting Aneurysmal Disease, HEALTH-F2-2008-200647).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Harakalova, M., van der Smagt, J., de Kovel, C. et al. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur J Hum Genet 21, 487–493 (2013). https://doi.org/10.1038/ejhg.2012.206
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.206
Keywords
This article is cited by
-
A harmful MYH11 variant detected in a family with thoracic aortic dissection and patent ductus arteriosus
Forensic Science, Medicine and Pathology (2023)
-
Association of gene polymorphisms in MYH11 and TGF-β signaling with the susceptibility and clinical outcomes of DeBakey type III aortic dissection
Mammalian Genome (2022)
-
The Genetics of Inheritable Aortic Diseases
Current Cardiovascular Risk Reports (2022)
-
Basement membrane collagen IV deficiency promotes abdominal aortic aneurysm formation
Scientific Reports (2021)
-
Epidemiology, presentation and population genetics of patent ductus arteriosus (PDA) in the Dutch Stabyhoun dog
BMC Veterinary Research (2016)

{kind=link}