
Abstract
X-linked retinal dystrophies (XLRD) are listed among the most severe RD owing to their early onset, leading to significant visual loss before the age of 30. One-third of XLRD are accounted for by RP2 mutations at the Xp11.23 locus. Deletions of ca. 1.2 Mb in the Xp11.3-p11.23 region have been previously reported in two independent families segregating XLRD with intellectual disability (ID). Although the RD was ascribed to the deletion of RP2, the ID was suggested to be accounted for by the loss of ZNF674, which mutations were independently reported to account for isolated XLID. Here, we report deletions in the Xp11.3-p11.23 region responsible for the loss of ZNF674 in two unrelated families segregating XLRD, but no ID, identified by chromosomal microarray analysis. These findings question the responsibility of ZNF674 deletions in ID associated with X-linked retinal dystrophy.
Similar content being viewed by others


Introduction
X-linked retinal dystrophies (XLRD) account for about 5–20% of families with RD.1, 2, 3, 4 Two genes, RP25 (MIM300757) and Retinitis pigmentosa GTPase regulator6, 7 (MIM312610), account for at least 80–90% of XLRD.4, 8
In 1994, Aldred et al9 reported a large kindred cosegregating XLRD and intellectual disability (ID). Subsequently, affected patients were found to carry a 1.27-Mb microdeletion in Xp11.3-p11.23 and the disease was regarded as a contiguous gene deletion syndrome.10 Another 1.2-Mb deletion in Xp11.3-p11.23 was later reported in a sporadic case of XLRD with ID.11 Both deletions encompassed exons 1–3 of RP2, the SLC9A7 (MIM300368), CHST7 (MIM300375), ZNF673 (MIM300585) and ZNF674 (MIM300573) genes, and two genes encoding highly conserved microRNAs (mir221 (MIM300568), mir222 (MIM300569)). Although the retinal degeneration was ascribed to the disruption of RP2 in both families, ID was suspected to result from the loss of one or more genes of the interval; ZNF674, in which mutations were identified in some patients affected with non-syndromic ID, was regarded as a strong candidate gene.11 Here, we report large Xp11.3-p11.23 deletions in two unrelated families segregating non-syndromic X-linked cone-rod dystrophy.
Clinical evaluation
Two unrelated families were ascertained through the Genetic Department at Necker Hospital in Paris. Written informed consents were obtained from the participants of the study.
In Family 1, the propositus (IV6, Figure 1) presented with high myopia at the age of 1. At the age of 3 years, macular and peripapillary retinal atrophy were evidenced at the fundus. At the age of 5 and half years, the atrophy had spread to the peripheral retina. Visual acuity was reduced to 4/200 (right eye (RE) and left eye (LE)). Electroretinographic (ERG) recordings evidenced severe alteration of both photopic and scotopic responses. The patient's psychomotor development is normal since birth. Today, at the age of 12, he has been enrolled in a school for the blind.

Pedigree and haplotypes at the RP2 locus of the two families segregating Xp11.3 deletions with non-syndromic X-linked retinal dystrophy. d: deleted. Base position according to UCSC Genome Browser GRCh37/hg19 assembly.
The review of the ophthalmological data revealed that affected male relatives (III6, III7, III9, IV9 and IV10; Figure 1) displayed very similar age and mode of onset of the disease, which is early severe myopia, macular rearrangements with preservation of the peripheral retina, but flat ERG responses before the age of 6. The disease worsened rapidly (before the age of 12) with reduced caliber of the retinal vessels, pigmentary deposits in the peripheral retina, night blindness and residual visual acuity <20/200 by the age of 30. They all presented with normal psychomotor development and were enrolled in normal middle schools before enrolling in high schools for the blind.
In Family 2, the propositus (III7, Figure 1) presented with jerk nystagmus, high bilateral myopia (−13.5doRE, −12.75doLE with astigmatism −2.5doLRE), diffuse retinal pigment epithelium atrophy at the fundus and normal ERG recordings at the age of 4 and half years old. Visual acuity was not recorded at this age. At the age of 8, fundus examination showed a bull's eye aspect of the macular region with peripapillary atrophy, peripheral atrophic retinal pigment epithelium with some pigmentary deposits and thin retinal vessels (Figure 2). Visual-field recordings displayed a central scotoma and a constricted peripheral visual field. Visual acuity was severely reduced (4/200 RE; 20/200 LE). Photopic and scotopic ERG responses were severely altered. The patient presented with normal psychomotor development since birth. Currently, at the age of 8, he is enrolled in a normal school. His maternal uncle (II4, Figure 1) developed normally. He presented with a high myopia since birth, but we were unable to obtain any detailed clinical data.

Ophthalmoscopic phenotype of Patient III7 (Family 2). (a) Fundus images showing peripapillary chorioretinal atrophy extending to the posterior pole, thin retinal vessels in the periphery, and some osteoblasts-like deposits. (b) Autofluorescence images showing a ‘bull-eye’ aspect of the macular region.
The ophthalmological data recorded in patients of the two families and summarized in Table 1 were consistent with the diagnosis of X-linked cone-rod dystrophy.
Methods and results
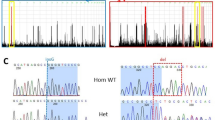
Linkage analysis using markers encompassing the RP2 and RP3 loci revealed absence of amplification with markers located close to the RP2 gene in male patients of both families, suggesting the presence of deletions (Figure 1). To characterize the boundaries of the predicted deletions, DNA of affected and unaffected males were subjected to PCR, using primers designed to amplify a series of STS markers specific to ZNF673, ZNF674, RP2 and PHF16 that lie in the DXS8083–GATA160B08 interval (Table 2; gene order tel-ZNF673–ZNF674–RP2–PHF16-cen). All PCRs resulted in positive amplification signals for unaffected male individuals of both families. Conversely, some STS markers could not be amplified from the genomic DNA of affected patients, allowing the positioning of the deletion breakpoints between the 5′-end of ZNF673 and the 3′-end of PHF16, and between the 3′-region of ZNF673 and exon 5 of RP2 in families 1 and 2, respectively (Figure 3, Table 2). Quantitative multiplex PCR of short fluorescent fragments12 identified the deletion in two non-obligate carriers (IV3 and IV8, Family 1; not shown, available on request).

Comparative genomic hybridization profiles of chromosome Xp11.3–p11.23, using high-resolution oligonucleotide microarray (Affymetrix Cytogenetics Whole-Genome 2.7 M Array) and PCR analysis of the region. Patient IV6 – Family 1 (upper panel) harbors a large deletion encompassing a 509-Kb sequence flanked by the C1DCNH and C1DZO probes. Patient III7 – Family2 (lower panel) harbors two close deletions: (i) a telomeric 431-Kb deletion flanked by the C1DCFA and C1DCM3 probes containing no known gene and (ii) a 388-kb centromeric deletion flanked by the C1DCNZ and C1DCX6 probes. The deleted regions in both patients included ZNF674, but not mir221 and mir222. The results of the array comparative genomic hybridization are consistent with the PCR analysis. The sequence and position of PCR amplicons a–q are presented in Table 2. +: Positive PCR amplification signal; −: negative PCR amplification signal. Structural genomic variations of the Xp11.3–p11.23 region are shown; inversions are in purple, whereas CNVs and indels resulting in a gain in size or a loss in size are in blue and red, respectively.
DNA samples of affected individuals IV6 (Family 1) and III7 (Family 2) were further analyzed using comparative genomic hybridization on a high-resolution oligonucleotide microarray (Affymetrix Cytogenetics Whole-Genome 2.7-M Array, Affymetrix UK Ltd, High Wycombe, UK). Calculation of test over reference Log2 intensity ratios identified a 509-Kbp deletion in Patient IV6 (Family 1) DNA, which proximal and distal deletion breakpoints mapped between probes C1DCNH and C1DCNJ (chromosome interval 46 317 965–46 320 660), and C1DCZN and C1DCZ0 (chromosome interval 46 830 359–46 831 436), respectively.
The deletion encompassed sequences between the 3′-end of ZNF673 and the 5′-half of PHF16 (Figure 3). Positions of the deletion boundaries were consistent with those suggested by PCR analysis of the STSs of the region (Figure 3; Table 2).
Patient III7 (Family 2) carried two neighboring deletions, encompassing 431 and 388 Kbp, respectively (Figure 3). Proximal and distal breakpoints of the centromeric deletion mapped between probes C1DCNZ and C1DCO0 (chromosome interval 46,336,830–46,350,109), and C1DCX4 and C1DCX6 (chromosome interval 46,738,728–46,740,394 intervals), respectively. The deletion encompassed sequences between the 3′-UTR of ZNF673 and RP2 intron 4 (Figure 3). These data were consistent with the PCR analysis of the STSs of the region (Figure 3; Table 2). Proximal and distal breakpoints of the telomeric deletion, which encompassed no known gene, mapped between probes C1DCFA and C1DCFB (chromosome interval 45 794 008–45 794 890), and C1DCM2 and C1DCM3 (chromosome interval 46 226 847–46 229 422), respectively. This result was confirmed by PCR analysis using primers designed to amplify STS markers flanking the deletion breakpoints (Figure 3; Table 2).
Gene order and probe positions are those of UCSC Genome Browser GRCh37/hg19 assembly (http://genome.ucsc.edu/).
Discussion
The ophthalmological phenotype of the patient harboring a Xp11.3-p11.23 microdeletion reported by Lugtenberg et al11 was poorly described. Conversely, the phenotype of the male patients in both families (Table 1) is consistent with the atypical phenotype of macular and peripapillary retinal atrophy reported earlier on a patient carrying a 4-bp deletion in the RP2 gene responsible for absent protein,13 and to that of patients harboring a 1.2-Mb deletion of the Xp11.3-p11.23 region described by Aldred et al.9 Together, these data suggest a strong correlation between this particular phenotype and the loss of RP2, which mutations usually account for rod-cone dystrophy.
In both families, all male patients (n=9, information available in 8/9) presented with normal psychomotor development since birth, despite the loss of the coding sequence of the ZNF674 gene. This observation raises the question of the relevance of the deletion of ZNF674 in the ID of patients cosegregating this feature with XLRD. Yet, it is worth noting that the Xp11.3-p11.23 deletions in these patients encompassed two highly conserved microRNAs – mir221 and mir222 – which were not deleted in the patients with isolated RD we report here. The two mirRNAs belong to the same cluster and they are not involved in structural genomic variation to our knowledge (Figure 3). They have an important role in cell cycle checkpoint that ensures cell survival by being involved in the coordination of cell proliferation.14 Both of them are expressed in the adult brain.15 During early fetal life, they are highly expressed in the cortex and cerebellum when neuron terminal differentiation is abundant.16 As the brain develops, they exhibit decreasing expression in the cortex and increasing expression in the cerebellum, in which growth appears later compared with the cortex and continues after birth.16 These data suggest that mir-221 and mir-222 have a role in the regulation of brain development. Interestingly, through the screening of all known brain-expressed X-chromosomal mirRNAs in a cohort of 464 patients with non-syndromic X-linked ID, only four nucleotide changes were identified in three mirRNAs, of which two out of four lie within the mir222 and segregated with the ID in two independent families.17 Although, it was not proven that these two changes underlie the disease, it was suggested that they might affect the Drosha cleavage site of the pre-mir222 and consequently might be able to influence the processing of its RNA products. Eventually, it is reasonable to consider that the ID in patients reported by Aldred et al9 and Lugtenberg et al11 may be accounted for by the loss of mir222, with or without that of mir221.
That being said, one should remember that the screening of the ZNF674 gene in 28 XLID families and 309 index patients with previous family history suggestive of X-linked inheritance resulted in the identification of a nonsense and two missense mutations in three patients, respectively.11 The absence of ID associated with the deletion of the ZNF674 gene is not inconsistent with the implication of ZNF674 point mutations in X-linked non-syndromic ID. These mutations may act in a dominant manner, including the nonsense mutation p.E118X.11 Indeed, the p.E118X mutation, which lies in the last exon of the gene, was present in patients’ mRNA, suggesting that the aberrant transcript is not prone to nonsense-mediated mRNA decay and may be translated into a protein lacking the 462 carboxy-terminal amino acids, with a similar structure as that of the predicted ZNF673 protein, although 54 amino acids shorter. This truncated protein may not be capable of DNA binding due to the loss of the zinc finger domains, but might still bind to its co-repressor KAP-1 and interact with the N-CoR complex. In this context of dominant ZNF674 point mutations, preferential inactivation of mutant X chromosomes would explain the absence of clinical expression in mothers. Interestingly, in the family segregating the p.E118X mutation, all five obligate carriers presented with skewed X-inactivation.11
In conclusion, other families are needed to determine the exact role of the ZNF674 gene in the occurrence of non-syndromic and syndromic ID, as well as the potential role of mirRNA close to this gene and deleted in the Xp11.3 syndrome, but not in the two families reported here.
References
Bird AC : X-linked retinitis pigmentosa. Br J Ophthalmol 1975; 59: 177–199.
Fishman GA : Retinitis pigmentosa. Genetic percentages. Arch Ophthalmol 1978; 96: 822–826.
Kaplan J, Bonneau D, Frézal J, Munnich A, Dufier JL : Clinical and genetic heterogeneity in retinitis pigmentosa. Hum Genet 1990; 85: 635–642.
Hartong DT, Berson EL, Dryja TP : Retinitis pigmentosa. Lancet 2006; 368: 1795–1809.
Schwahn U, Lenzner S, Dong J et al: Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet 1998; 19: 327–332.
Meindl A, Dry K, Herrmann K et al: A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet 1996; 13: 35–42.
Roepman R, van Duijnhoven G, Rosenberg T et al: Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum Mol Genet 1996; 5: 1035–1041.
Pelletier V, Jambou M, Delphin N et al: Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum Mutat 2007; 28: 81–91.
Aldred MA, Dry KL, Knight-Jones EB et al: Genetic analysis of a kindred with X-linked mental handicap and retinitis pigmentosa. Am J Hum Genet 1994; 55: 916–922.
Zhang L, Wang T, Wright AF et al: A microdeletion in Xp11.3 accounts for co-segregation of retinitis pigmentosa and mental retardation in a large kindred. Am J Med Genet 2006; 140A: 349–357.
Lugtenberg D, Yntema HG, Banning MJG et al: ZNF674: a new Kruppel-associated box-containing zinc-finger gene involved in nonsyndromic X-linked mental retardation. Am J Hum Genet 2006; 78: 265–278. (Note: Erratum: Am J Hum Genet 2006; 78: 897.).
Casilli F, Di Rocco ZC, Gad S et al: Rapid detection of novel BRCA1 rearrangements in high-risk breast-ovarian cancer families using multiplex PCR of short fluorescent fragments. Hum Mutat 2002; 20: 218–226.
Dandekar SS, Ebenezer ND, Grayson C et al: An atypical phenotype of macular and peripapillary retinal atrophy caused by a mutation in the RP2 gene. Br J Ophthalmol 2004; 88: 528–532.
Medina R, Zaidi SK, Liu CG et al: MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res 2008; 68: 2773–2780.
Olsen L, Klausen M, Helboe L, Nielsen FC, Werge T : MicroRNAs show mutually exclusive expression patterns in the brain of adult male rats. PLoS One 2009; 4: e7225.
Podolska A, Kaczkowski B, Busk PK et al: MicroRNA expression profiling of the porcine developing brain. PLoS One 2011; 6: e14494.
Chen W, Jensen LR, Gecz J et al: Mutation screening of brain-expressed X-chromosomal miRNA genes in 464 patients with nonsyndromic X-linked mental retardation. Eur J Hum Genet 2007; 15: 375–378.
Acknowledgements
We thank the Association Retina France for its support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Delphin, N., Hanein, S., Taie, L. et al. Intellectual disability associated with retinal dystrophy in the Xp11.3 deletion syndrome: ZNF674 on trial. Guilty or innocent?. Eur J Hum Genet 20, 352–356 (2012). https://doi.org/10.1038/ejhg.2011.217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.217
Keywords
This article is cited by
-
Clinical findings and a DNA methylation signature in kindreds with alterations in ZNF711
European Journal of Human Genetics (2022)
-
Mapping the landscape of tandem repeat variability by targeted long read single molecule sequencing in familial X-linked intellectual disability
BMC Medical Genomics (2018)
