
Abstract
Screening for carriers of spinal muscular atrophy (SMA) is necessary for effective clinical/prenatal diagnosis and genetic counseling. However, a population-based study of SMA prevalence in mainland China has not yet been conducted. In this study, the copy number of survival motor neuron (SMN) genes was determined in 1712 newborn cord blood samples collected from southern China and from 25 core families, which included 26 SMA patients and 44 parents, to identify SMA carriers. The results presented 13 groups with different SMN1/SMN2 ratios among 1712 newborn individuals, which corresponded to 1535 subjects with two copies of SMN1, 119 with three copies of SMN1, 17 with four copies of SMN1, and 41 with a heterozygous deletion of SMN1 exon 7. Simultaneously, two ‘2+0’ genotypes and two point mutations were found among the 44 obligate carriers in the core families, including a novel SMN1 splice-site mutation that was identified in the junction between intron 6 and exon 7 (c. 835–1G>A). These results indicated that the carrier frequency is 1/42 in the general Chinese population and that duplicated SMN1 alleles and de novo deletion mutations are present in a small number of SMA carriers. In addition, we developed and validated a new alternative screening method using a reverse dot blot assay for rapid genotyping of deletional SMA. Our research elucidated the genetic load and SMN gene variants that are present in the Chinese population, and could serve as the basis for a nationwide program of genetic counseling and clinical/prenatal diagnosis to prevent SMA in China.
Similar content being viewed by others

INTRODUCTION
Spinal muscular atrophy (SMA) is one of the most common autosomal recessive disorders worldwide: the carrier frequency is between 1/40 and 1/60 in diverse populations and it affects 1 in 10 000 live births.1, 2, 3, 4 SMA is caused by the degeneration of anterior horn cells of the spinal cord, which leads to symmetric proximal muscle weakness and atrophy.5 On the basis of the age at onset and the clinical course, SMA is classically subdivided into three types: type I is an infantile acute form, and shows the greatest severity; type II is an infantile chronic form with intermediate severity; and type III is a childhood and adolescent form and shows the mildest severity.6
All three types of SMA are linked mostly to the survival motor neuron (SMN) gene at 5q13, which is found as two nearly identical copies: SMN1 and SMN2.7 Mutations in the telomeric copy of the SMN gene (SMN1) are responsible for SMA, whereas SMN2 functions as a disease-modifying gene that can ameliorate the clinical severity of SMA in a copy-dependent manner. Each SMN gene encompasses 27 kb and comprises 9 exons (exons 1, 2a, 2b, and 3–8). Typically, the DNA sequence of SMN1 differs from that of SMN2 by only five nucleotides: one in intron 6, one in exon 7, two in intron 7, and one in exon 8.8, 9 Of the SMA patients whose disorder is linked to 5q13, ∼96% are characterized by a homozygous deletion of SMN1 exon 7, which results from unequal crossover or SMN1-to-SMN2 conversion events. The remaining 4% of cases are caused either by compound heterozygosity with a point mutation in one allele and a deletion in the other or by compound heterozygous point mutations in the SMN1 gene.10 To date, ∼40 subtle mutations have been identified in the SMN1 gene of SMA patients worldwide, with the most common sites of mutation located in exons 3 and 6.11, 12
Given that homozygous deletion of exon 7 of SMN1 is a common genotype and SMN2 can influence the severity of SMA, it is fundamentally important to directly determine the copy numbers of SMN1 and SMN2 for carrier screening and clinical/prenatal diagnosis of SMA. Using quantitative analysis of SMN1/SMN2 gene dosage, a range of molecular investigations of the incidence of carriers and the genetic burden of SMA in diverse ethnic populations have been carried out.2, 3, 13, 14, 15, 16, 17, 18, 19 However, a population-based study of SMA prevalence in mainland China has not yet been executed. Although some data have been reported for subjects from Taiwan and Hong Kong, the analyses included a relatively small number of SMA patients.20, 21 To estimate the future burden of SMA and the requirements for its appropriate treatment, management, and prevention in the Chinese population, accurate population frequency data are essential. Such data could be obtained through the genotyping of SMN genes in a large number of samples from mainland China.
In the study reported herein, we determined SMN copy number in 1712 samples of newborn cord blood from southern China and from 25 core families that contained individuals with SMA using a modified denaturing high-performance liquid chromatography (DHPLC)-based quantitative assay, which has been proven to be a highly efficient and reliable carrier-screening test.20, 22, 23 We also assessed the genotype contribution of SMA carriers in the Chinese population by comparative genotyping of newborn samples and SMA core families. In addition, during the analysis of SMA patients, we found a novel SMN1 splice-site mutation at the junction between intron 6 and exon 7 (c.835–1G>A). Simultaneously, we developed and validated a new alternative method that used a reverse dot blot (RDB) assay for rapid genotyping of deletional SMA in our sample cohorts. We anticipate that our research will provide a basis for a nationwide program of clinical/prenatal diagnosis to prevent SMA in China.
MATERIALS AND METHODS
Population and patient samples
A total of 1712 cord blood samples obtained from healthy newborns (867 males and 845 females), with no family history of SMA and registered parental origin from the northern and southern districts of Guangdong province, were continuously collected from July 2006 to December 2007. Of these individuals, 95% were ethnically southern Chinese Han. In addition, 25 unrelated SMA core families from across China were enrolled in this study, including 26 SMA patients and 44 parents (2 affected siblings in one family), comprised of 19 parental pairs and 6 single parents. All patients fulfilled the diagnostic criteria of the International SMA Consortium.6 Genomic DNA was extracted from the peripheral blood of the core family members or from newborns’ cord blood. The local medical research ethics committee reviewed the study, and informed consent was obtained from all participants.
DHPLC analysis of SMN copy number in the tested samples
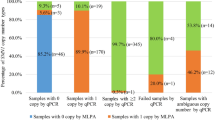
The diagnostic flow chart for determining SMN1 and SMN2 gene dosages linked to SMA in this study is illustrated in Figure 1. This protocol includes a single PCR to generate amplified segments containing exon 7 of SMN1/SMN2, followed by DHPLC quantitative analysis to display the SMN1/SMN2 ratio representing genotypic patterns of samples on the WAVE Nucleic Acid Fragment Analysis System (Model 4500 HT, Transgenomic, Omaha, NE, USA). To test the samples for an equal SMN1/SMN2 ratio (1:1 or 2:2), multiplex PCRs were performed to amplify SMN1/SMN2 by simultaneously amplifying two reference genes (X-linked CYBB and KRIT1 on 7q), followed by DHPLC quantitative analysis to distinguish the SMN1/SMN2 ratio as 1:1 or 2:2. We could clearly differentiate SMN1 and SMN2 PCR fragments of the same size by the DHPLC elution pattern, which is dependent on a single-nucleotide polymorphism (SNP) in exon 7 (C → T) present in these two genes. All primers used in this essay were described in detail by Su et al20 The reaction mixture components and PCR/DHPLC conditions were similar to the description by Su et al with minor revisions. All of the genomic DNA from the tested individuals were subjected to single PCR/DHPLC analysis to determine carriers with an SMN1/SMN2 ratio not equal to 1 (SMN1/SMN2=1:4, 1:3, and 1:2), whereas the normal individuals (accounting for nearly 40% of the total sample) were characterized by an unequal SMN1/SMN2 ratio (SMN1/SMN2=2:1, 2:3, 3:2, 3:1, and 4:1) or by the detection of an SMN1-only peak (SMN1/SMN2=2:0, 3:0, and 4:0). Samples with an equal SMN1/SMN2 ratio were then analyzed by the multiplex PCR/DHPLC assay to distinguish carriers with an equal SMN1/SMN2 ratio (1:1) from normal individuals with an equal SMN1/SMN2 ratio (2:2). Finally, all samples with abnormal DHPLC peaks were subjected to direct PCR sequencing to detect any novel SMN1/SMN2 gene mutations, and the SNP functional prediction and alignment analyses were performed according to the programs found at http://compbio.cs.queensu.ca/F-SNP/, whereas SMN1 and SMN2 gene dosages were determined for the diagnosis of individuals as either carrier or normal individuals. To analyze SMA patients with homozygous deletions from core families, SMA-affected cases were detected efficiently simply by recognizing an SMN2-only DHPLC peak.

Diagnostic flow chart for targeted detection of SMN1 and SMN2 gene dosages linked to SMA in a population-based study. The sample numbers detected at each step are indicated in brackets. *Some samples with abnormal peaks screened by DHPLC were sequenced to distinguish point mutations or SNPs.
Development and validation of the RDB assay to genotype the SMA deletion
We established RDB analysis to detect SMA deletions according to a previously described RDB system.24 The two pairs of primers labeled with biotin to selectively amplify the exon 7 and exon 8 regions of the SMN1 (Genebank NG: 008691) and SMN2 (Genebank NG: 008728) genes were as follows: biotin-AGACTATCAACTTAATTTCTGATCA-3′ (from 31 844 to 31 868 bp) and biotin-TAAGGAATGTGAGCACCTTCCTTC-3′ (from 32 022 to 32 045 bp); biotin-GTAATAACCAAATGCAATGTGAA-3′ (from 32 666 to 32 688 bp) and biotin-CTACAACACCCTTCTCACAG- 3′ (from 32 835 to 32 855 bp), respectively. The two sets of oligonucleotide probes immobilized on strips were designed to distinguish between the SMN1 and SMN2 genes. The four specific hybridization probes, which were linked to the amino group at the 5′-end, were SMN1-Ex7: NH2-TGTCTGAAACCCTGTA-3′ (from 31 996 to 32 011 bp), SMN2-Ex7: NH2-TGTCTAAAACCCTGTAAG-3′ (from 31 992 to 32 0095 bp), SMN1-Ex8: NH2-AAAGACTGGGGTGGG-3′ (from 32 726 to 32 740 bp), and SMN2-Ex8: NH2-AAAGACTGAGGTGGG-3′ (from 32 724 to 32 738 bp). Multiplex PCR amplification was performed, and the 50 μl total reaction mixture contained 100 ng of gDNA, 0.5 μ M of each primer for exon 7, 0.4 μ M of each primer for exon 8, 400 μ M of each dNTP, 4 Units of Taq polymerase (Takara, Dalian, China), and 5 μl of 10 × buffer I. The cycle conditions used for PCR were 5 min at 94°C for denaturation, followed by 35 cycles of 45 s at 94°C, 30 s at 55°C, 30 s at 72°C, and a final extension of 10 min at 72°C. The conditions used for hybridization, washing, and color development in this RBD assay were as described in our previously published paper24. The RDB assay was validated using 183 samples (which were blinded with regard to the previous diagnosis) by DHPLC/DNA sequencing, including 25 samples with homozygous deletions of exon 7 and/or exon 8 of the SMN1 gene and 158 samples with an SMN1/SMN2 ratio that derived from normal or SMA carrier individuals. The DHPLC/DNA sequencing results were unknown until the genotypes obtained from each of the given RDB results were scored.
Molecular identification of a novel mutation in a Chinese family
The proband was obtained from an 8-month-old male infant from 1 of the 25 SMA families who was clinically diagnosed as SMA type I, without the homozygosity typical of SMN1 gene deletion. The patient was referred to us for further molecular analysis. Blood samples were collected from the patient, his elder sister, and his parents for identification with the new mutation and functional study with SMN1 RNA expression and abnormal RNA spliceosome. DHPLC, followed by direct DNA sequencing, was used to identify the unknown mutation in the SMN1 gene. Total RNA was isolated from peripheral blood lymphocytes using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). First-strand cDNA synthesis and reverse transcription were performed according to the manufacturer's instructions (Fermentas Fermentas, Hanover, MD, USA) using 30 ng each of the forward primer (5′-ACCACCACCCCACTTACTA-3′, specific for exon 5 of SMN1/SMN2) and the reverse primer (5′-TTTTTCTTTCCCAACACTTGAT-3′, specific for exon 8 of SMN1/SMN2). The reaction mixture components and PCR conditions were identical to those described for the above single PCR reaction used before the DHPLC assay.
Statistical analyses
Current and predicted vital statistics were obtained from the ‘National Population and State Family Planning Commission of China’.25 Statistical analyses were conducted using SPSS 17.0 statistical software (SPSS Inc., Chicago, IL, USA). Differences in the allele/genotype frequency of SMN1 genes were calculated from the modified Hardy–Weinberg formula.
RESULTS
SMN1/SMN2 copy number and population prevalence
Characterization of the SMN1/SMN2 ratio or copy numbers in 1712 newborn individuals by DHPLC analysis resulted in the identification of 13 different groups. We identified 41 subjects with a heterozygous deletion of SMN1 exon 7, which indicated that the carrier prevalence was 2.39% in the general Chinese population. The number and percentage of subjects with each different SMN1/SMN2 ratio among our samples are listed in Table 1, and the frequencies of the SMN1 alleles and genotypes defined by this study are listed in Table 2. Representative DHPLC profiles for different SMN1/SMN2 peak ratios are shown in Figure 2. From the results of the DHPLC analysis, we identified a group of subjects who seemed to carry novel SMN1/SMN2 gene mutations. Seven SNPs in the introns or exons of the SMN1 or SMN2 gene were identified in 31 subjects by direct sequencing; this included 20 subjects with mutation IVS6–50A>G, 6 with IVS6–24dupT, 4 with IVS7+287dupC, 2 with IVS7+137G>T, 2 with IVS7+183C>T, 1 with c.875C>G, and 1 with EX8+204del2. These seven variants of the SMN1 and SMN2 genes could not be associated with causative factors of SMA in the Chinese population on the basis of data obtained from SNP functional prediction and alignment analysis. No deviation from Hardy–Weinberg and genotypic equilibrium was observed by testing the allele and genotype distributions for the SMN1/SMN2 loci.

Representative DHPLC profiles of single/multiplex PCR analysis for different SMN1/SMN2 ratios. *Female individuals with SMN1/SMN2=1:1;**male individuals with various SMN1/SMN2 ratios.
Characterization of the SMA genotypes of the core families
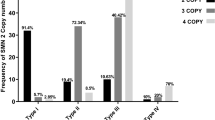
Among the 26 SMA patients from the 25 unrelated core families, 13 cases of type I, 5 cases of type II, and 8 cases of type III SMA were detected (Table 3). In all cases, at least 1 copy of the SMN1 gene was deleted: 24 were homozygous deletions of SMN1, and 2 were heterozygous deletions. The SMN1/SMN2 copy numbers in the 44 carriers from these SMA families were determined by DHPLC analysis and compared with the copy numbers obtained for the 41 carriers in the newborn population (Table 4). No significant differences in the genotype frequencies of SMA carriers were observed between these two groups (P>0.05). As expected, the ‘1+0’ genotype was predominant, and accounted for 90.9% (40/44) of carriers in the SMA core families and 100% (41/41) in the newborn population. Among three carriers from the core families with an SMN1/SMN2 ratio of 2:1, we found that the fathers of two unrelated patients had the ‘2+0’ genotype and we also identified two ‘1+1D’ genotypes in individuals with an SMN1/SMN2 ratio of 2:2 or 2:1.
Development and validation of the RDB assay
The RDB assay was developed to genotype deletions of SMN1 exon 7 and exon 8 using a panel of samples from SMA patients, carriers, or normal individuals who had been characterized previously by molecular analysis. Figure 3 shows representative RDB profiles for four tested samples. Results indicate that the assay could clearly distinguish SMA patients who carried a homozygous deletion (only SMN2 could be detected) from normal individuals or carriers with only SMN1 genes or with different ratios of SMN1/SMN2. The assay could not discriminate a normal individual from a carrier with different ratios of SMN1/SMN2 genes. In a blinded experiment, the results for 182 of the 183 samples (99.5%) tested by the RDB assay were in agreement with the results obtained by DHPLC/DNA sequencing; the exception was the misdiagnosis of one sample that contained one of the below-mentioned novel mutations.

Representative results for RDB detection of different SMN1/SMN2 ratios. (a) Typing of a normal individual with SMN1/SMN2=2:N; (b) typing of an SMA carrier with SMN1/SMN2=1:N; (c) typing of a normal individual with SMN1 only; (d) typing of SMA patients with SMN1/SMN2=0:N.
A novel SMN1 splice-site mutation
A novel acceptor splice-site mutation (c.835–1G>A) within intron 6 of the SMN1 gene was discovered in 1 of the 25 unrelated core families using DHPLC, RDB, and DNA sequence analysis (Figure 4). The proband had inherited a typical deletion of SMN1 exon 7 from his mother and a novel mutation from his father. DNA sequencing revealed unique peak heights at the c.835–1G>A mutation (Figure 4e) in the genomic DNA of both the proband and his father and confirmed the results obtained by the DHPLC quantitative assay, which showed that the ratio of SMN1/SMN2 was 2:2 in the father and 1D:3 in the proband. In addition, the sequences of SMN1 and SMN2 vary at nucleotide position +6 in exon 7 (C in SMN1, T in SMN2). Figure 4e shows that the peak size of C was equal to that of T for the father, whereas the peak size of C was one-third that of T for the proband, which further confirmed that the ratio of SMN1/SMN2 was 2:2 in the father and 1D:3 in the proband. RT-PCR analysis of RNA from the father demonstrated the expected pattern of SMN1/SMN2 expression: the abundance of the 294-bp product (SMN1 Δexon7 allele and SMN2) was much greater than that of the 348-bp product (SMN1 wild-type allele). The 294-bp product represents amplification of both the SMN2 gene transcript, from which exon 7 is normally deleted, and the SMN1 transcript with the splice-site mutation (c.835–1G>A), which indicates that the mutation inhibits splicing of the SMN1 gene.

SMN analysis for a family carrying the novel SMN1 splice-site mutation c.835–1G>A. (a) Pedigree and SMN copy numbers for the patient's family. (b) RT-PCR amplification of SMN gene exons 5–8 to determine SMN1/ SMN2 expression in the RNA sample obtained from the proband's father. The transcript product for Δexon7 was 294 bp and the normal transcript product including SMN1 exon 7 was 348 bp in length. M: DNA molecular weight marker; lanes 1 and 2: normal individuals; lanes 3 and 4: the father carried the SMN1 splice-site mutation at the junction between intron 6 and exon 7 (c.835–1G>A). (c) RDB analysis of the SMN gene in the proband and his father. The DNA fragment hybridized with the probe for SMN1 exon 7 contained the site with the splice-site mutation c.835G>A, which provided a negative result by the RDB assay for the proband (panel b). However, the father was an SMA carrier with one normal SMN1 gene, which provided a positive result by the RDB assay (panel a). (d) Quantitative DHPLC analysis of the SMN1/SMN2 gene dosage in the proband and his father. (Panel a) The father exhibited a gene ratio of SMN1/SMN2=2:2; (panel b) the proband exhibited a gene ratio of SMN1/SMN2=1:3. (panel e) DNA sequencing of the SMN1 gene carried by the proband and his father. (Panel a) Sequence of genomic DNA containing SMN gene intron 6 and exon 7 from the father; (panel b) sequence of the genomic DNA containing SMN gene intron 6 and exon 7 from the proband. The downward arrow indicates a G → A mutation at the acceptor splice site of intron 6.
DISCUSSION
We studied the incidence of carriers and genetic background of SMA in mainland China by quantifying the copy numbers of SMN1 and SMN2 genes in 1712 newborn individuals using a DHPLC-based assay. This is the first large-scale population-based genetic investigation of SMA in the general population of mainland China. We selected random individuals from the southern Han Chinese population as representative of the population of mainland China, because the two groups of Chinese, the southern Han Chinese and the northern Han Chinese, who represent the largest population in mainland China share a common origin and similar genetic structure.26, 27 Nearly all of the possible different ratios of SMN1/SMN2, as determined by quantitative DHPLC-based assay, could be observed clearly in the Chinese population. This confirmed the complex structural organization of the human SMN gene and is similar to previous observations in Caucasian, Korean, Australian, and African populations.2, 3, 13, 14, 15, 16, 17, 18, 19 Simultaneously, we compared SMN1 alleles and genotype frequencies among different races. There were no significant differences among Chinese, Caucasian, Korean, Australian, American, and African populations, which showed that there were no racial or regional differences with respect to SMA in general populations. As expected, the ‘1+0’ genotype was predominant among carriers, and accounted for 90.9% (40/44) of the carriers in the SMA core families and 100% (41/41) in the newborn population, but it should be noted that a few distinct, rare genotypes (2+0 and 1+1D) were neglected in our newborn cohort because they could not be detected by the present assay. In addition, among three carriers from the core families with an SMN1/SMN2 ratio of 2:1, we found that the fathers of two unrelated patients had the ‘2+0’ genotype and we also identified two ‘1+1D’ genotypes in individuals with an SMN1/SMN2 ratio of 2:2 or 2:1. Given that the number of copies of the SMN2 gene is undoubtedly the major modifier of the SMA phenotype, the elucidation of the SMN2 gene patterns presented herein will provide fundamental knowledge of this disorder in the Chinese population. These data demonstrate a carrier prevalence of 1/42 (2.39%) in the general Chinese population with respect to heterozygosity for deletion and intragenic mutations of SMN1 exons 7 and 8; the infrequent SMN gene defects that consist of point mutations outside of exons 7 and 8 were not detected. In addition, the ‘2+0’ genotype could not be identified in this study. The data provide a reliable representation of the incidence of SMA carriers in the general Chinese population because analysis of the allele and genotype distributions for the SMN1/SMN2 loci showed no deviation from Hardy–Weinberg equilibrium. In addition, the ‘1+0’ genotype was proven to be predominant in the Chinese population on the basis of data obtained from the SMA core family study (Table 4). Therefore, our results could provide a basis for a nationwide program of genetic counseling, clinical screening, and prenatal diagnosis to prevent SMA in China. According to the expected frequency of homozygous deletion of SMN1 in newborns (Table 2), 14.34 individuals in 100 000 (95% CI, 13.66/100 000∼15.03/100 000) will have the ‘0+0’ genotype. With a current annual birth rate of ∼16 080 000,25 we can estimate that the number of newborns in mainland China who will be affected by SMA is 2306 (95% CI, 2196∼2417) per year.
Individuals with the rare ‘2+0’ genotype have two copies of SMN1 in cis on one chromosome and no SMN1 gene on the other chromosome, which results in a compound SMA heterozygous deletion with a carrier frequency that ranges from 1.7 to 3.3% in different ethnic populations.1, 3, 13 In the family study, we detected two cases of the ‘2+0’ genotype among the 44 parents of the SMA patients; these parents had an SMN1/SMN2 ratio of 2:1 (Table 3 and 4). This finding suggests that the ‘2+0’ genotype is a considerable SMA determinant and should be tested during carrier screening and clinical molecular diagnosis in the Chinese population. Population estimations suggest that approximately two (1.87) ‘2+0’ carriers should be present among the 1712 newborns analyzed but presumably they were not detected because of false-negative results obtained in our DHPLC assay. Direct detection of the ‘2+0’ SMA carriers still remains a great challenge because of the time-consuming and laborious detection methods required. Therefore, the finding of two normal dosages of the SMN1 gene may significantly reduce the risk of being a carrier, but there is still a small risk of recurrence among future offspring for individuals with two SMN1 gene copies. Given that it is a rare event and there will be a relatively high number of samples with an SMN1/SMN2 ratio of 2:1, we suggest that it would be more economical and efficient to focus molecular analysis of the ‘2+0’ genotype on index patients and their family members rather than implementing a population-wide screening. On the other hand, the fact that 2% of individuals with SMA have one de novo mutation, and a few individuals have a rare point mutation in the SMN1 gene,28, 29 might provide an explanation of possible false-negative results when interpreting the SMN1 gene dosage-based test for individuals undergoing genetic counseling. Moreover, Bayesian analysis is essential for the risk assessment calculations that are required for the proper genetic counseling of families with a history of SMA.
By using two groups of samples, the genotype of which had been characterized by the DHPLC assay, we have established and validated a new, simple, and reliable RDB method that is capable of directly detecting the most common homozygous deletion of SMN1 in exons 7 and 8. This methodology has desirable features that include its ease of operation and the lack of a requirement for expensive equipment or strict experimental conditions, which makes it suitable for use in different laboratories and is especially beneficial for developing countries. According to our experience, it can be used as an alternative method to genotype the SMA homozygous deletion directly in routine clinical diagnosis. The current assay was limited to the detection of this common deletion mutation. We intend to extend the assay to include other relatively rare point mutations so that the assay would be capable of detecting all types of SMA mutations in the Chinese population in the future.
In conclusion, our research has determined the genetic load and the SMN gene variants that are present in the Chinese population, and these results should serve as the basis for a nationwide program of genetic counseling, population screening, and prenatal diagnosis to prevent SMA in China. We have also proven that the DHPLC-based dosage analysis is a suitable method to determine the carrier status of individuals with a family history of SMA or to investigate cases of clinically suspected SMA with a negative diagnostic SMA test. However, it should be noted that reasonable counseling modalities must be adapted to different individuals, as described by Smith et al3 Given that the DHPLC-based dosage analysis does not detect all carriers, especially those with the ‘2+0’ genotype or intragenic SMN mutations, a risk assessment is an essential component for clinical screening of individuals who have two or more copies of SMN1. Genetic risk assessments can be estimated principally on the basis of our reported carrier frequency for the ‘1+0’ genotype. However, the ‘2+0’ genotype, de novo mutations, and rates of intragenic mutations in the SMN1 gene are additional factors that need to be considered. Furthermore, as demonstrated for the Australian population, a multimodal approach that involves SMN dosage analysis, linkage analysis, and genetic risk assessment facilitates the resolution of SMA carrier status in individuals with a family history, as well as among individuals in the general population.3 Such an approach should provide Chinese couples who are undergoing genetic counseling with improved choices for their family planning. In addition, we have successfully validated the RDB assay as an alternative method for the rapid genotyping of deletional SMA in routine clinical diagnoses.
References
Ogino S, Wilson RB : Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet 2002; 111: 477–500.
Cusin V, Clermont O, Gerard B et al: Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet 2003; 40: e39.
Smith M, Calabro V, Chong B et al: Population screening and cascade testing for carriers of SMA. Eur J Hum Genet 2007; 15: 759–766.
Ogino S, Wilson RB, Gold B : New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet 2004; 12: 1015–1023.
Monani UR : Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 2005; 48: 885–896.
Munsat TL, Davies KE : International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromusc Disord 1992; 2: 423–428.
Scharf JM, Endrizzi MG, Wetter A et al: Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat Genet 1998; 20: 83–86.
Lefebvre S, Bürglen L, Reboullet S et al: Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165.
Monani UR, Lorson CL, Parsons DW et al: A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999; 8: 1177–1183.
Wirth B : An update on the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 2000; 15: 228–237.
Wirth B, Brichta L, Hahnen E : Spinal muscular atrophy: from gene to therapy. Semin Pediatr Neurol 2006; 13: 121–131.
Zapletalová E, Hedvicáková P, Kozák L et al: Analysis of point mutations in the SMN1 gene in SMA patients bearing a single SMN1 copy. Neuromuscul Disord 2007; 17: 476–481.
Feldkotter M, Schwarzer V, Wirth R et al: Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Lee TM, Kim SW, Lee KS et al: Quantitative analysis of SMN1 gene and estimation of SMN1 deletion carrier frequency in Korean population based on real-time PCR. J Korean Med Sci 2004; 19: 870–873.
Basel-Vanagaite L, Taub E, Drasinover V et al: Genetic carrier screening for spinal muscular atrophy and spinal muscular atrophy with respiratory distress 1 in an isolated population in Israel. Genet Test 2008; 12: 53–56.
Ogino S, Leonard DG, Rennert H et al: Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet 2002; 110: 301–307.
McAndrew PE, Parsons DW, Simard LR et al: Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 1997; 60: 1411–1422.
Corcia P, Mayeux-Portas V, Khoris J et al: Abnormal SMN1 gene copy number is a susceptibility factor for amyotrophic lateral sclerosis. Ann Neurol 2002; 51: 243–246.
Anhuf D, Eggermann T, Rudnik-Schoneborn S et al: Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum Mutat 2003; 22: 74–78.
Su YN, Hung CC, Li H et al: Quantitative analysis of SMN1 and SMN2 genes based on DHPLC: a highly efficient and reliable carrier-screening test. Hum Mutat 2005; 25: 460–467.
Chan V, Yip B, Yam I et al: Carrier incidence for spinal muscular atrophy in southern Chinese. J Neurol 2004; 251: 1089–1093.
Chen WJ, Wu ZY, Lin MT et al: Molecular analysis and prenatal prediction of spinal muscular atrophy in Chinese patients by the combination of restriction fragment length polymorphism analysis, denaturing high-performance liquid chromatography, and linkage analysis. Arch Neurol 2007; 64: 225–231.
Shaw SW, Cheng PJ, Chang SD et al: Rapid prenatal diagnosis of spinal muscular atrophy by denaturing high-performance liquid chromatography system. Acta Obstet Gynecol Scand 2008; 87: 960–968.
Li L, Zhou YQ, Xiao QZ et al: Development and evaluation of a reverse dot blot assay for the simultaneous detection of six common Chinese G6PD mutations and one polymorphism. Blood Cells Mol Dis 2008; 41: 17–21.
Official web site for National Population and State Family Planning Commission of China; (http://www.gdpic.gov.cn/).
Su B, Xiao C, Deka R et al: Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Hum Genet 2000; 107: 582–590.
Wen B, Li H, Lu D et al: Genetic evidence supports demic diffusion of Han culture. Nature 2004; 431: 302–305.
Wirth B, Schmidt T, Hahnen E et al: De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997; 61: 1102–1111.
Cusco I, Barcelo MJ, del Rio E et al: Detection of novel mutations in the SMN Tudor domain in type I SMA patients. Neurology 2004; 63: 146–149.
Acknowledgements
We sincerely thank Dr Yi-Ning Su (Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan) for his help and advice with the DHPLC method and Professor Feng-hua Lan (Fuzhou General Hospital of Nanjing Military Area, Fuzhou) for providing part of the SMA core family samples. We also thank all the participants for providing samples. This work was partially supported the Ministry of Science and Technology of China for the National Key Technologies R & D Program (2006BAI05A06) and the Genetic Resource R & D Program (30771192) and by the National Science Fund for Distinguished Young Scholars (30325037).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sheng-Yuan, Z., Xiong, F., Chen, YJ. et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur J Hum Genet 18, 978–984 (2010). https://doi.org/10.1038/ejhg.2010.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.54
Keywords
This article is cited by
-
Dual Mechanism of a New SMN1 Variant (c.835G>C, p.Gly279Arg) by Interrupting Exon 7 Skipping and YG Oligomerization in Causation of Spinal Muscular Atrophy
Journal of Molecular Neuroscience (2021)
-
NGS-based spinal muscular atrophy carrier screening of 10,585 diverse couples in China: a pan-ethnic study
European Journal of Human Genetics (2021)
-
High-throughput screening reveals novel mutations in spinal muscular atrophy patients
Italian Journal of Pediatrics (2020)
-
Transmission characteristics of SMN from 227 spinal muscular atrophy core families in China
Journal of Human Genetics (2020)
-
Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review
Orphanet Journal of Rare Diseases (2017)
