
Abstract
The X-linked creatine transporter defect is caused by mutations in the SLC6A8 gene. Until now, 66 synonymous and intronic variants in SLC6A8 were detected in our laboratory. To gain more insight in the effect of the detected variants, we applied five free web-based splice-site analysis tools to 25 published variants that were stratified as (non-)disease causing. All were correctly predicted to have no effect (n=18) or to cause erroneous splicing (n=7), with the exception of a pathogenic de novo 24 bp intronic deletion. Second, 41 unclassified variants, including 28 novel, were subjected to analysis by these tools. At least four splice-site analysis tools predicted that three of the variants would affect splicing as the mutations disrupted the canonical splice site. Urinary creatine/creatinine and brain MRS confirmed creatine transporter deficiency in five patients (four families), including one female. Another variant was predicted to moderately affect splicing by all five tools. However, transient transfection of a minigene containing the variant in a partial SLC6A8 segment showed no splicing errors, and thus was finally classified as non-disease causing. This study shows that splice tools are useful for the characterization of the majority of variants, but also illustrates that the actual effect can be misclassified in rare occasions. Therefore, further laboratory studies should be considered before final conclusions on the disease-causing nature are drawn. To provide an accessible database, the 109 currently known SLC6A8 variants, including 35 novel ones, are included in a newly developed LOVD DNA variation database.
Similar content being viewed by others

Introduction
In 2001, the X-linked mental retardation (XLMR) syndrome, SLC6A8 deficiency, was identified because of a creatine deficiency in the brain caused by mutations in the creatine transporter (SLC6A8) gene (MIM 300036). SLC6A8 has been mapped to Xq281 and is a member of the solute-carrier family 6 (neurotransmitter transporters). The clinical presentation of males affected with SLC6A8 deficiency is mental retardation (MR), expressive speech and language delay, epilepsy, developmental delay and autistic behavior. Laboratory hallmarks include a reduction of the creatine signal in the proton magnetic resonance spectroscopy (H-MRS) of the brain, an increased urinary creatine/creatinine ratio and impaired creatine uptake in cultured fibroblasts. In female carriers, learning disabilities of varying degrees have been noted. Two studies of males with XLMR estimated the prevalence of SLC6A8 deficiency to be 2.1% (CI: 0.44–3.76)2 and 1.5% (CI: 0–4.46).3 A third study of males with MR revealed a prevalence of 0.8% (CI: 0.02–1.7%).4 Within the last decade many novel variants in the SLC6A8 gene have been detected. These variants are difficult to classify as pathogenic or non-disease causing because the variants are either located in the coding region, but are synonymous, or are found in the intronic regions. However, such variants may affect proper splicing and are potentially pathogenic.5 We therefore not only tested if such variants could be properly analyzed by studying previously classified variants, but also subjected 28 novel variants to these tools. Moreover, to facilitate worldwide diagnostic and research laboratories interested in the SLC6A8 gene, we developed a novel LOVD database (http://www.LOVD.nl/SLC6A8), which includes clinically and genetically relevant data.
Materials and methods
Subjects
In our diagnostic unit, a total of 1900 patients with a differential diagnosis of SLC6A8 deficiency were analyzed by DNA sequence analysis. This resulted in the detection of 66 individuals with intronic or synonymous variants (including 28 novel). These variants are addressed in this study. Also, five novel patients affected with SLC6A8 deficiency are reported.
Methods
PCR of exon 1–13 of SLC6A8 (NM_005629.1)
Exon 2 to 13 and flanking intronic sequences of SLC6A8 were amplified using HotStarTaq Polymerase (Qiagen, Valencia, CA, USA). Amplification consisted of an initial denaturation step at 95 °C for 15 min, followed by 38 cycles of 94 °C for 45 s, 66 °C for 45 s and 72 °C for 80 s. For the amplification of exon 1 and its flanking sequences, Takara LA Taq (Takara Bio Inc., Otsu, Shiga, Japan) Polymerase was used. After an initial denaturing step for 1 min at 94 °C, amplification was allowed in 35 cycles composed of 95 °C for 30 s, 66 °C for 30 s and 72 °C for 80 s.
RT-PCR analysis of SLC6A8 cDNAs and minigenes
RNA was isolated from lymphoblasts or fibroblasts using the SV RNA kit (Promega, Madison, WI, USA) or from PAX blood tubes (Qiagen). cDNA was synthesized from isolated RNA using Omniscript Reverse Transcriptase (Qiagen) according to the manufacturer's instructions. RT-PCR was performed using cDNA primers designed for the exons of interest of the SLC6A8 gene. To rule out possible amplification of genomic DNA, RT-PCR for each RNA was also performed without reverse transcriptase.
Construction and transfection of the minigenes
Owing to the large size of the SLC6A8 gene and its highly GC-rich 5′region, we only cloned the region of interest of the SLC6A8 gene. As templates for the PCR, the patients' and a wild-type genomic DNAs were used. The fragment, covering exons 3–7 including 53 and 55 nucleotides of the flanking 5′ and 3′ intronic regions respectively, was amplified with forward primer 5′-CCGGAATTCGTAAAACGACGGCCAGCAGGGGGAGGTGGCCAGGG-3′ containing an EcoR1 site and reverse primer 5′-GCGTCGACCAGGAAACAGCTATGACATGCATCTGGGTAGCACTC-3′ with a SalI restriction site for cloning into the pBABE-puro plasmid. The fragment was amplified using Takara LA Taq (Takara Bio Inc.), cloned into the TOPO-TA (Invitrogen, Breda, The Netherlands) vector and either wild-type or mutant sequence was confirmed by sequencing. For the construction of the positive control (ie, c.777+2T>A), site directed mutagenesis was performed on the TOPO vector containing the wild-type fragment with forward primer 5′-AAATCCACGGGAAAGGAACCACTAGAGGCATGC-3′ and reverse 5′-GCATGCCTCTAGTGGTTCCTTTCCCGTGGATTT-3′. Presence of the desired mutation and absence of PCR-artifacts were confirmed by sequencing of the complete fragments. After digestion with EcoR1 and Sal1, the inserts were sub cloned into the pBABE-puro plasmid.
Minigene constructs were transiently transfected into SLC6A8-deficient primary fibroblasts. With the use of polyethylenimine (PEI), 25 μg of construct, in a ratio of 3:1 (PEI:DNA), was transfected in fibroblasts grown to 70% confluence in a 75 cm2 culture disc. After 48 h, cells were harvested.
DNA sequence analysis
Sequence analysis was performed using BigDye v3.1 terminator and an ABI 3130xl (Applied Biosystems, Nieuwerkerk aan de IJssel, The Netherlands). The obtained sequences were analyzed using the Mutation Surveyor software package (Softgenetics, State College, PA, USA).
Analysis of amplicons with DHPLC
For DHPLC analysis, optimal heteroduplex formation was realized by denaturing PCR products at 95 °C for 5 min and gradual cooling to 25 °C over a period of 60 min. Subsequently, 5 μl of the heteroduplex/homoduplex mixture was loaded on a WAVE 3500HT DNA fragment analysis system (Transgenomic, Omaha, NE, USA). Elution of the PS-DVB DNAsep column (Transgenomic) was performed in high-throughput mode with a linear gradient of increasing acetonitril (ACN) concentration with a runtime of 3 min and a constant flow rate of 1.5 ml/min. Gradient was realized with the use of buffer A (0.1 M triethylammonium acetate (TEAA) and 0.025% ACN) and buffer B (0.1 M TEAA and 25% ACN). Navigator 2.0 software (Transgenomic) was used to calculate the ratio of buffer A and B during the gradient, as well as the optimal partially denaturing temperature (Tm) for the amplicon. The Tm of this amplicon was determined as 64 °C.
In silico analysis
The analysis of splicing efficiencies in the normal and mutant sequences was carried out using the following five splice-site analysis tools, which are commonly used in diagnostic laboratories: the Berkeley Drosophila Genome Project6 (http://www.fruitfly.org/seq_tools/splice.html), Netgene27 (http://www.cbs.dtu.dk/services/NetGene2/), Splice Predictor8 (http://deepc2.psi.iastate.edu/cgi-bin/sp.cgi), GenscanW9 (http://genes.mit.edu/GENSCAN.html) and FSplice (http://linux1.softberry.com/berry.phtml?topic=fsplice&group=programs&subgroup=gfind). The parameters used for analysis were the default settings of all tools.
LOVD database
The target of the LOVD database is to include all published and unpublished mutations and variants. It is localized at a server in Leiden, The Netherlands, which can be reached by the following URL http://www.LOVD.nl/SLC6A8 or through the Variation Databases page of the Human Genome Variation Society (HGVS, www.HGVS.org/). The SLC6A8-specific database has been developed with the recently described LOVD software,10 which is in agreement with the HGVS guidelines.11 All variants are checked and approved by Mutalyzer.12 So far 38 pathogenic mutations have been reported in 44 male patients affected with SLC6A8 deficiency.
Results
Validation of splice-site analysis using previously reported variants
We analyzed seven pathogenic mutations (Table 1) detected in patients affected with SLC6A8 deficiency by applying five splice-site analysis tools (see methods). In six out of seven pathogenic mutations at least four of the five tools predicted a reduced recognition of the splice sites, varying with a reduction of the probability score between 9 and 100%. Three mutations had significant reduction scores of >99% in four of the five tools, whereas three other variants had reduction of their scores in a much lower range. One mutation, c.1392+24_1393-30del, reducing the intron from 76 to 52 bp, was not recognized by all the tools. Although analysis of this mutation with Splice Predictor resulted in the loss of both rho and gamma values, this was not ascribed to the mutation itself but to Splice Predictor's precondition of an intron size of at least 60 bp.
In addition, 18 previously reported variants that are not disease causing were correctly predicted not to cause erroneous splicing by all five splice-site analysis tools, with the exception of one synonymous variant that showed in four tools minor reduction scores of equal or <7%. These variants are all confirmed to be non-disease causing, because we now either established at the cDNA level that no erroneous splicing occurred or that the variants were detected in healthy control males or were published in dbSNP (see Table 1; see www.lovd.nl/slc6a8).
Detection and classification of novel variants
Within the last decade, we have analyzed about 1900 individuals (referred to our department because of, eg, MR, increased urinary creatine/creatinine) by DNA sequence analysis of the SLC6A8 gene to exclude/confirm creatine transporter defect. In this endeavor, we identified 41 variants including 28 novel synonymous and intronic variants that were not included in the dbSNP database and of which their (non-) disease-causing nature needed to be established. All were subjected to the five splice-site analysis tools. In total, three novel pathogenic mutations (c.263-1G>C, c.778-2A>G and c.1596+1G>A) were detected that were predicted to cause aberrant splicing with reduction scores of 100% by at least three splice-site analysis tools (Table 2). Twenty-seven variants showed no effect by all five tools. By only one or two tools eight variants were predicted to decrease the possibility of correct splicing by 10% or less. One synonymous unclassified variant (c.780C>T) showed in three splice-site analysis tools reduction scores that appear significant in terms of percentage, but if the normal range is taken into account these differences are considered minimal (eg, the score by the FruitFly tool shows a decrease in 32% while the actual score decreases from 0.22 to 0.15 with the range of low probability to high probability being 0–1.00). All these variants were considered non-disease causing. Only one intronic unclassified variant (c.777+4C>T) was predicted by all five splice-site analysis tools to reduce the probability of the canonical donor site of exon 4 (Table 2) by 10, 2, 23, 10 and 15% compared with the canonical donor site score. cDNA was not available from this patient and thus this variant was studied by overexpression of a minigene (ie, a genomic SLC6A8 segment containing the c.777+4C>T variant). Both overexpression of the wild-type minigene as well as the mutant minigene in SLC6A8-deficient primary fibroblasts showed normal splicing as detected by RT-PCR and sequence analysis. This is in contrast to the positive control minigene that contained the c.777+2T>A mutation and resulted in skipping of exon 4 (Figure 1).

RT-PCR results of spliced products after transfection of the minigenes in SLC6A8-deficient fibroblasts. SLC6A8-deficient fibroblast were transfected with a minigene, containing an exon 3 to 7 fragment of SLC6A8 with either a wild-type sequence, the c.777+2T>A transversion or the c.777+4C>T variant. Cells were harvested and RNA was isolated using the Promega RNA Isolation Kit. Subsequent RT-PCR of the wild-type and the c.777+4C>T minigene transfected fibroblasts resulted in an amplicon of 384 bp, indicating the variant not to cause aberrant splicing. The positive control (c.777+2T>A transfectants) resulted in a 251 bp amplicon. Sequencing analysis showed that exon 4 was skipped in the 251 bp amplicon.
Five novel SLC6A8-deficient patients
The first case (patient I) was a young female (DOB January 1993) born at term following a normal pregnancy and delivery with normal weight, length and head circumference. From the age of 2, she developed a behavior disturbance and was assessed by a pedopsychiatric institution that also diagnosed mild MR. At age 14, she was affected with auditory hallucinations and the diagnosis of chronic hallucinatory psychosis was made. Her cognitive impairment was stable with conserved language capacities and social contact, no motor impairment and no specific neurological signs. Her total IQ was tested at 45 (verbal: 61, reason: 45, memory: 53; and working velocity: 66). A brain H-MRS was performed which showed a reduced cerebral creatine level with a normal appearance to choline and N-acetylaspartic acid (Figure 2). Urinary creatine levels were within the upper normal range. Plasma creatine was determined twice resulting in values of 39.9 μmol/l and 75.4 μmol/l (normal 30–124 μmol/l). Genomic DNA sequencing revealed a c.263-1G>C mutation. This mutation arose de novo. Somatic mosaicism was not detected in her parents' DNA by both DNA analysis and the use of DHPLC (Figure 3). mRNA isolated from PAX tubes, followed by cDNA synthesis showed approximately 90% r.263_325del (p.Gly88_Leu108del) and 10% wild-type product. The X-inactivation was studied in blood with the analysis of HhaI digested and undigested DNA followed by PCR of the highly polymorphic CAG repeat of the androgen receptor gene and showed a 90:10 pattern. Also, fibroblasts incubated with a physiological concentration of creatine (25 μ M) showed no uptake. All these findings combined confirmed creatine transporter deficiency.

Proton MRS of the brain of an index girl with SLC6A8 deficiency (patient I). A proton MRS was performed on a 14-year-old girl with mild mental retardation. Creatine (Cr) was found to be reduced with a normal appearance to choline (Cho) and N-acetylaspartic acid (NAA). Subsequent genomic DNA sequencing revealed a pathogenic c.263-1G>C mutation, which confirmed the diagnosis of SLC6A8 deficiency.

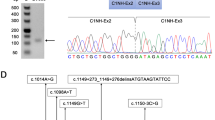
Patient I with a c.263-1G>C mutation in the SLC6A8 gene. Somatic mosaicism for the c.263-1G>C mutation was not detected in both parents of patient I. The DHPLC elution profile of the exon 2 amplicon of EDTA blood DNA of patient I with a c.263-1G>C variant in SLC6A8 including her parents and a male control. At the retention time of the heteroduplex peak clearly visible with the patient, no peak is visible with the parents, indicating both are not somatic mosaic for the mutation.
The second case (patient II) was a 7-year-old male with epilepsy, expressive language difficulties and a movement disorder. He was tested for mutations in SLC6A8 after H-MRS of the brain revealed a diminished creatine peak with a normal appearance to NAA and choline and urinary analysis showed an increased Cr/Crn value. This resulted in the detection of a hemizygous c.778-2A>G variant. His brother (patient III), currently 10 years old, showed a similar clinical phenotype with an increased urinary Cr/Crn value and absence of cerebral creatine. He was also found to be hemizygous for this variant while his mother was proven to be heterozygous.
The fourth patient (patient IV) was a young male with a reduced cerebral creatine level measured by H-MRS. Further investigation resulted in the detection of a c.1596+1G>A variant, which was also found to be heterozygous in DNA from his mother.
The fifth patient (patient V) was unmistakably diagnosed with SLC6A8 deficiency at the age of 6 years. The patient suffered from moderate MR and severe language delay. Quantitative localized single voxel magnetic resonance spectroscopy was performed over the basal ganglia and revealed a markedly diminished creatine level with a normal appearance to choline. Urinary analysis revealed a creatine/creatinine value of 1.7 (normal 0.017–0.72).13 The creatine uptake ability of the patients' fibroblasts was significantly decreased in comparison with control cells when incubated at a physiological creatine concentration of 25 μ M. Moreover, the mutation could not be detected in the DNA of the mother and is therefore considered de novo. At the cDNA level, two erroneous transcripts were revealed (r.[1392_1393ins1392+1_1393-1;1392+24_1393-30del,1393_1495del]) (Figure 4). This conclusively classified the c.1392+24_1393-30del mutation as the pathogenic event. Also, the same de novo mutation was detected in an unrelated patient.14

Schematic representation of the c.1392+24_1393-30del variant in SLC6A8. The c.1392+24_1393-30del variant was found in two independent patients and occurred de novo in both cases. In the patients' DNA a hemizygous variant, c.1392+24_1393-30del, was found (upper pane). mRNA analysis showed that this variant affects splicing and results in different splice products, r.[1392_1393ins1392+1_1393-1;1392+24_1393-30del, 1393_1495del] (lower pane), resulting in SLC6A8 deficiency. For the second patient see Hathaway et al.14 This variant is annotated according to the guidelines of Den Dunnen and Antonarakis (http://www.genomic.unimelb.edu.au/mdi/mutnomen/).
LOVD database
In total, the LOVD database of SLC6A8 lists 44 SLC6A8-deficient families, with a total of 43 mutations. Of these, 38 are proven pathogenic (+/+), whereas 5 others are presumed to be pathogenic (+?/+?). The latter are all missense variants, which have not been investigated by overexpression studies yet.15
The database (Figure 5) provides detailed information on the nature of the variants, but also exon–intron location of the DNA change (both according to current nomenclature and as published), RNA change and protein change. To all variants a unique database number is assigned. The column ‘variant remarks’ provides a detailed description on the nature of the variant. The final conclusion is presented in the first column ‘path.’ in which the first sign provides the conclusion of the report and the second one that of the curator (eg, +/+ meaning confirmed pathogenic). Other columns include detailed information on the variant origin, reference, template, technique, frequency, disease status, patient remarks, times reported, gender, geographic origin and ethnic origin. Moreover, clinical symptoms are continuously archived (non-public). The database is an excellent tool to get an up-to-date overview of the type of mutations, the frequency of specific variants and more details. This data can be found in the different tabs by any user, but an advanced search function is also available. Researchers or clinicians who are interested in the nature of a specific variant can use the database to search if the variant has been detected in other patients. This information may have been published in reports or on the website only, but also may include unpublished variants, which have not yet been made public. The latter allows contact between clinicians even if the data are not publicly released. All scientists/clinicians are invited to submit their data to the database. The identity of the submitter is included in the database.

Screenshot of the newly developed LOVD/SLC6A8 database. The currently known variants and pathogenic mutations of SLC6A8 are described in the newly developed LOVD database including the corresponding clinical data. Search queries can be performed for many parameters (ie, specific variant, exon number, biochemical findings). In order to submit variants it is required to log in, after which the variants will be checked for correctness and completeness by the curator.
Discussion
In this study, the nature of 67 intronic and synonymous variants has been studied by free web-based splice-site analysis tools. These have been characterized and are all being included in a newly developed LOVD database. In addition, all previously published mutations are included in this database as well. This is of high relevance for researchers and clinicians who detect difficult to interpret variants in the SLC6A8 gene. In an ideal diagnostic setup, an unclassified variant is further investigated at the mRNA, protein and/or functional level to confirm/exclude its pathogenic nature. However, in diagnostic laboratories time is usually limited and proper materials are often not available. Therefore, alternative methods are warranted for variant classification such as frequently used splice-site analysis tools, which predict the possible effect on RNA splicing of a variant based on known and computed conserved splicing sequences. Here we demonstrate that the use of five splice-site analysis tools, indeed is very helpful for proper classification.
In total, 24 out of 25 (96%) variants were properly classified by the combined use of the five tools. The importance of using more than one splice-site analysis tool is illustrated by the fact that four out of seven proven pathogenic mutations were not predicted to have a strong effect (ie, 90% or higher reduction) by one or more of the individual analysis tools, whereas the combined data of all the tools clearly predicted erroneous splicing for six of these variants (Table 1). In one case, only one tool predicted a reduction in the probability of correct splicing. The fact that this ‘missed’ mutation (c.1392+24_1393-30del) comprises an intronic deletion of 24 bp out of a 76 bp intron, easily explains this omission as none of the five splice-site analysis tools have intron size as a parameter. This caveat is an important finding for diagnostic laboratories that encounter this specific type of mutations (eg, deletions, insertions in small introns). We demonstrated that this deletion indeed results in erroneous spliced transcripts (r.[1392_1393ins1392+1_1393-1;1392+24_1393-30del,1393_1495del]), (Hathaway et al,14 this study).
In 17 out of 18 proven variants, no change in prediction scores was detected by any of the five tools. Only one variant that was proven not to interfere with proper splicing, had a reduced score of 7% or less compared with the canonical site by four of the five tools. Owing to this variant, we arbitrarily decided to only consider variants for further molecular workup if at least three tools predicted a mild effect (10<>80%), or if one or two tools predicted a significant effect (>80%). In addition, we took into account the prediction score of the wild-type splice site. Thus, a 50% reduction of a canonical site with a 0.99 score is more likely to have an effect on splicing than a 50% reduction in a splice site with a much weaker score of for instance 0.22.
Utilizing these criteria, we analyzed the potential effect of 41 unclassified variants on the SLC6A8 mRNA with the use of these five splice-site analysis tools. This resulted in the identification of three pathogenic mutations (c.263-1G>C, c.778-2A>G and c.1596+1G>A), and 38 variants that have no apparent relation with creatine transporter deficiency. The effect on splicing for two of the three novel pathogenic mutations could not be confirmed at the cDNA level. However, given the fact that all three mutations affect the donor or acceptor splice-site, all were classified as pathogenic. Pathogenicity was in excellent agreement with the fact that the mutations were not detected in 280 control chromosomes, the biochemical findings (increased urinary Cr/Crn) and/or reduced cerebral creatine detection by H-MRS.
Interestingly, one novel mutation (c.263-1G>C) was found in a girl, who according to our knowledge is the first reported index girl affected with SLC6A8 deficiency. The girl was not correctly diagnosed until the age of 14 when a brain H-MRS revealed a reduced cerebral creatine level. This led to DNA analysis that resulted in the discovery of the pathogenic c.263-1G>C mutation. Additional investigations revealed a urinary creatine to creatinine ratio within the normal range as well as normal values for plasma creatine. The discrepancy between the urinary, plasma and cerebral creatine levels is possibly caused by variation of the X-inactivation pattern in different tissues, which was shown to be skewed in blood of this patient.
In a recently described cohort of female relatives who were heterozygous for a pathogenic mutation in SLC6A8, it was shown that symptoms of SLC6A8 deficiency (eg, MR, learning difficulties and constipation) can occur in female heterozygotes. However, it should be noted that the level of biochemical markers (urinary creatine to creatinine ratio and/or cerebral creatine) usually overlap with that of normal controls. This is in concordance with the clinical and biochemical findings of the above mentioned female index patient and confirms the recommendation of screening for mutations in SLC6A8 in females with (mild) MR/psychiatric disorders as this method appears to be the most sensitive and specific test.16
In only 1 of the 38 UVs, mild reduction values of the probability scores were detected by all five splice-site analysis tools. Unfortunately, it was not possible to obtain additional material of the patient to isolate mRNA, and clinically it was also not possible to exclude/confirm SLC6A8 deficiency. We therefore selected this variant (c.777+4C>T) for overexpression studies of a minigene containing the variant. These studies revealed no aberrant splicing and therefore this variant is also considered non-pathogenic.
After evaluating the results of the splice-site analysis tools reported in this study, we defined our criteria for the analysis of SLC6A8. If at least three out of five splice-site analysis tools predict a reduction of >10% or one out of five predicts a reduction of 80% of the site score compared with the canonical site, further research is warranted. However, a critical view needs to be kept which is illustrated by the fact that in our study 1 out of the 10 proven pathogenic mutations would have escaped proper identification. The c.1392+24_1393-30del mutation was not recognized by the overall prediction score of all five splice-site analysis tools. It is noteworthy that these scores do not include composition, size and branch site of the intron as parameters. One exception is Splice predictor, which does have additional intron-dependent scores, namely rho and gamma. Unfortunately, these scores are not calculated and set at zero when an intron is smaller than 60 bp, therefore making it difficult to deduce the actual reduction with this variant as the deletion leads to a 52 bp intron. This of course reduces the reliability of these scores because naturally occurring introns can have sizes of 60 bp and less.
In conclusion, splice-site analysis tools are very important in the process of classifying novel variants. For definite classification of novel variants outside the canonical donor and acceptor sites, in vitro experimentation, either with mRNA analysis or with the use of a minigene (if no additional material is available), is an essential procedure.
References
Gregor P, Nash SR, Caron MG, Seldin MF, Warren ST : Assignment of the creatine transporter gene (SLC6A8) to human chromosome Xq28 telomeric to G6PD. Genomics 1995; 25: 332–333.
Rosenberg EH, Almeida LS, Kleefstra T et al: High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am J Hum Genet 2004; 75: 97–105.
Betsalel OT, van de Kamp JM, Martínez-Muñoz C et al: Detection of low-level somatic and germline mosaicism by denaturing high-performance liquid chromatography in a EURO-MRX family with SLC6A8 deficiency. Neurogenetics 2008; 9: 183–190.
Clark AJ, Rosenberg EH, Almeida LS et al: X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum Genet 2006; 119: 604–610.
Salomons GS, Bok LA, Struys EA et al: An intriguing ‘silent’ mutation and a founder effect in antiquitin (ALDH7A1). Ann Neurol 2007; 62: 414–418.
Reese MG, Eeckman FH, Kulp D, Haussler D : Improved splice site detection in Genie. J Comput Biol 1997; 4: 311–323.
Hebsgaard SM, Korning PG, Tolstrup N et al: Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res 1996; 24: 3439–3452.
Brendel V, Xing L, Zhu W : Gene structure prediction from consensus spliced alignment of multiple ESTs matching the same genomic locus. Bioinformatics 2004; 20: 1157–1169.
Burge C, Karlin S : Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997; 268: 78–94.
Fokkema IF, den Dunnen JT, Taschner PE : LOVD: easy creation of a locus-specific sequence variation database using an ‘LSDB-in-a-box’ approach. Hum Mutat 2005; 26: 63–68.
Den Dunnen JT, Antonarakis SE : Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 15: 7–12.
Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE : Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat 2008; 29: 6–13.
Almeida LS, Verhoeven NM, Roos B et al: Creatine and guanidinoacetate: diagnostic markers for inborn errors in creatine biosynthesis and transport. Mol Genet Metab 2004; 82: 214–219.
Hathaway SC, Friez M, Limbo K et al: X-Linked creatine transporter deficiency presenting as a mitochondrial disorder. J Child Neurol 2010; 25: 1009–1012.
Rosenberg EH, Martínez-Muñoz C, Betsalel OT et al: Functional characterization of missense variants in the creatine transporter gene (SLC6A8): improved diagnostic application. Hum Mutat 2007; 28: 890–896.
van de Kamp JM, Mancini GM, Pouwels PJ et al: Clinical features and X-inactivation in females heterozygous for creatine transporter defect. Clin Genet 2010, e-pub ahead of print 7 May 2010. doi:10.1111/j.1399-0004.2010.01460.x.
Hahn KA, Salomons GS, Tackels-Horne D et al: X-linked mental retardation with seizures and carrier manifestations is caused by a mutation in the creatine-transporter gene (SLC6A8) located in Xq28. Am J Hum Genet 2002; 70: 1349–1356.
Lion-Francois L, Cheillan D, Pitelet G et al: High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology 2006; 67: 1713–1714.
Mancini GM, Catsman-Berrevoets CE, de Coo IF et al: Two novel mutations in SLC6A8 cause creatine transporter defect and distinctive X-linked mental retardation in two unrelated Dutch families. Am J Med Genet A 2005; 132: 288–295.
Schiaffino MC, Bellini C, Costabello L et al: X-linked creatine transporter deficiency: clinical description of a patient with a novel SLC6A8 gene mutation. Neurogenetics 2005; 6: 165–168.
Wilcken B, Fagan E, Sim K, Carpenter KH, Salomons GS : Creatine transporter defect: results of 6 months treatment. J Inherit Metab Dis 2008; 31: 278P.
Acknowledgements
We thank Donald E Brass for his excellent reviewing. The work of Ofir T Betsalel, Efraim H Rosenberg, Ligia S Almeida and Gajja S Salomons is supported by the Dutch society for Scientific Research (ZonMW/NWO), VIDI grant number 917.56.349 and the Horst Bickel Award 2002, that of Tjitske Kleefstra by the European Union grant QLG3-CT-2002-01819 EURO-MRX, that of Johan T den Dunnen by the European Community's Seventh Framework Programme (FP/2007-2013) under grant agreement no. 200754 – the GEN2PHEN project and that of Charles Schwartz by grants of NICHD (HD26202) and the South Carolina Department of Disabilities and Special Needs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Betsalel, O., Rosenberg, E., Almeida, L. et al. Characterization of novel SLC6A8 variants with the use of splice-site analysis tools and implementation of a newly developed LOVD database. Eur J Hum Genet 19, 56–63 (2011). https://doi.org/10.1038/ejhg.2010.134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.134
Keywords
This article is cited by
-
A novel SLC6A8 mutation associated with intellectual disabilities in a Chinese family exhibiting creatine transporter deficiency: case report
BMC Medical Genetics (2018)
-
A novel SLC6A8 mutation associated with motor dysfunction in a child exhibiting creatine transporter deficiency
Human Genome Variation (2015)
-
Diagnostic methods and recommendations for the cerebral creatine deficiency syndromes
Pediatric Research (2015)
-
Carrier frequency of guanidinoacetate methyltransferase deficiency in the general population by functional characterization of missense variants in the GAMT gene
Molecular Genetics and Genomics (2015)
-
X‐linked creatine transporter deficiency: clinical aspects and pathophysiology
Journal of Inherited Metabolic Disease (2014)
