
Abstract
Aortic dilatation/dissection (AD) can occur spontaneously or in association with genetic syndromes, such as Marfan syndrome (MFS; caused by FBN1 mutations), MFS type 2 and Loeys–Dietz syndrome (associated with TGFBR1/TGFBR2 mutations), and Ehlers–Danlos syndrome (EDS) vascular type (caused by COL3A1 mutations). Although mutations in FBN1 and TGFBR1/TGFBR2 account for the majority of AD cases referred to us for molecular genetic testing, we have obtained negative results for these genes in a large cohort of AD patients, suggesting the involvement of additional genes or acquired factors. In this study we assessed the effect of COL3A1 deletions/duplications in this cohort. Multiplex ligation-dependent probe amplification (MLPA) analysis of 100 unrelated patients identified one hemizygous deletion of the entire COL3A1 gene. Subsequent microarray analyses and sequencing of breakpoints revealed the deletion size of 3 408 306 bp at 2q32.1q32.3. This deletion affects not only COL3A1 but also 21 other known genes (GULP1, DIRC1, COL5A2, WDR75, SLC40A1, ASNSD1, ANKAR, OSGEPL1, ORMDL1, LOC100129592, PMS1, MSTN, C2orf88, HIBCH, INPP1, MFSD6, TMEM194B, NAB1, GLS, STAT1, and STAT4), mutations in three of which (COL5A2, SLC40A1, and MSTN) have also been associated with an autosomal dominant disorder (EDS classical type, hemochromatosis type 4, and muscle hypertrophy). Physical and laboratory examinations revealed that true haploinsufficiency of COL3A1, COL5A2, and MSTN, but not that of SLC40A1, leads to a clinical phenotype. Our data not only emphasize the impact/role of COL3A1 in AD patients but also extend the molecular etiology of several disorders by providing hitherto unreported evidence for true haploinsufficiency of the underlying gene.
Similar content being viewed by others

Introduction
Aortic dilatation/dissection (AD) is a life-threatening condition associated with considerable morbidity and mortality. It can occur spontaneously, because of cardiovascular risk factors (eg, hypertension), or in association with genetic disorders, such as familial thoracic aortic aneurysms leading to type A dissections (TAAD, MIM #132900), Marfan syndrome (MFS, MIM #154700), Loeys–Dietz syndrome (LDS, MIM #609192), and the vascular type of Ehlers–Danlos syndrome (EDS IV, MIM #130050). MFS is an autosomal dominant systemic disorder of connective tissue (1–2:10 000).1 It shows variable manifestations in the cardiovascular, skeletal, and ocular systems.2 In the majority of cases, MFS is caused by mutations in the FBN1 gene (MIM *134797), which encodes the extracellular matrix protein fibrillin-1.3 The molecular etiology of MFS has been extended by the finding that heterozygous mutations in the genes encoding transforming growth factor-β receptors I (TGFBR1, MIM *190181) and II (TGFBR2, MIM *190182) can lead to MFS-related disorders, such as MFS type 2 (MFS2; MIM #610380), LDS, and TAAD.4, 5, 6, 7, 8
EDS IV, the vascular type of EDS, is an autosomal dominant disorder of connective tissue as well, but less prevalent than MFS (∼1–2:100 000).9 In addition to complications in the cardiovascular system (arterial rupture), manifestations of EDS IV involve the skin, joints, and hollow organs, resulting in thin/translucent skin, extensive bruising, characteristic facial appearance, and intestinal/uterine rupture as major diagnostic criteria.10 EDS IV is caused by mutations in COL3A1 (MIM *120180), which encodes the α1 chain of type III collagen.11, 12, 13, 14 So far, nearly 200 unique COL3A1 mutations have been registered in locus-specific mutation databases (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk; Leiden Open Variation Database, https://eds.gene.le.ac.uk), some of which led to functional haploinsufficiency by preferential degradation of mutant transcripts due to nonsense-mediated mRNA decay (NMD). However, no case of true COL3A1 haploinsufficiency, that is, complete loss of one allele through hemizygous deletion, has been described at the molecular level before (cf. Supplementary Table S1 and Supplementary Figure S1).
In this study, we report a hemizygous deletion of COL3A1 and flanking genes as well as assess the clinical and biochemical effects of this deletion. In addition to the importance of COL3A1 mutations in AD patients, our results show the different role of true haploinsufficiency in the etiology of dominant disorders.
Materials and methods
Patients
A total of 100 unrelated AD patients with familial (∼20/100) or sporadic (∼80/100) phenotypes suggestive for TAAD/MFS/LDS/EDS IV and thus in some cases also involving the skeletal (∼40/100) and/or ocular (∼5/100) system were selected for this study. In this cohort, previous sequencing and multiplex ligation-dependent probe amplification (MLPA) analyses of FBN1, TGFBR1, and TGFBR2 revealed no pathogenic sequence variation.8, 15, 16 Data on the clinical phenotypes were collected from medical records or during physical examinations by one of the authors. Informed consent was obtained from patients and family members, and the study was approved by the responsible local ethics committee.
Multiplex ligation-dependent probe amplification
MLPA was performed using 100 ng template DNA (referred to us or extracted from blood, tissue, or fibroblast samples) and the MLPA kit P155 (MRC-Holland, Amsterdam, The Netherlands), which contains MLPA probes for 10 of the 51 COL3A1 exons, according to the manufacturer's instructions. MLPA fragments were separated by capillary electrophoresis on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Rotkreuz, Switzerland). Each MLPA signal was normalized and compared with the corresponding peak area obtained in control DNA samples. Deviations of >30% were suspected as alterations and verified by repeated MLPA analysis.
High-density microarray analyses
To narrow down the breakpoints of the hemizygous deletion identified by MLPA in patient 53B, high-density microarray analyses were performed using the GeneChip Human Mapping 500 K Array Set (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's instructions. Data analysis was performed as described elsewhere.16
Breakpoint analyses
Based on decreased microarray signal intensities, primers flanking the predicted deletion were designed (P53B_7F 5′-AAAAATAGGGCAATGTCAACTAA-3′, P53B_17R 5′-CTCGACCAGCTTCAGAACT-3′) and used in long-range PCR. Accordingly, the Expand Long Template PCR System (Roche Diagnostics, Rotkreuz, Switzerland) was used with 100 ng of DNA, Buffer 3, and thermal cycling program (annealing for 30 s at 58 °C and elongation for 15 min at 68 °C) as described previously.16 Amplification products were sequenced using internal primers (P53B_7c_F 5′-GCAACAATGAATGGGAGAGA-3′, P53B_16c_R 5′-ACTCTGAATCAGCACCACTTG-3′) and standard procedures.16 Family members were tested by both MLPA and a PCR-based assay using primers designed for sequencing of the deletion breakpoints.
Biochemical testing and electron microscopy
Cultured dermal fibroblasts from the index patient 53B, his mother (53D), and two of his affected brothers (53 and 53E) were radiolabeled. Subsequently, collagens in medium and cell layer (harvested separately) were treated with pepsin, precipitated with ethanol, separated by SDS-PAGE (5%), and visualized by fluorography.17 In addition, for patients 53, 53D, and 53E, a portion of the skin biopsies was processed for transmission electron microscopy as reported.18 Furthermore, for patient 53E, his mother (53D), his daughter (53I), and his son (53J) as well as for his wife (53M), as a control subject, standard laboratory blood parameters thought to be associated with hemochromatosis type 4 were determined (for details, see Supplementary Table S2).
Statistical analysis
For proportions, the upper and lower limits of the 95% confidence interval were calculated.15
Results
Multiplex ligation-dependent probe amplification
We had screened 100 FBN1-, TGFBR1-, and TGFBR2-mutation-negative AD patients for large COL3A1 deletions/duplications by MLPA. In one of these patients (53B), the relative peak areas of all MLPA probes for COL3A1 were reduced, suggesting a hemizygous deletion of the entire gene (Supplementary Figure S2). The remaining 99 patients showed relative peak areas within the normal range. As the MLPA kit used in this study contains only probes for 10 of the 51 exons of COL3A1 (exons 1, 2, 5, 11, 17, 20, 29, 32, 43, and 51; NM_000090.3), in the 99 negatively tested patients only loss or gain of these 10 exons could be excluded. In addition, mosaicisms and copy-number neutral rearrangements cannot be excluded as such cases may not be detectable by MLPA. Thus, in patient cohorts comparable to that used in this study the relative frequency of deletions and duplications of the 10 analyzed COL3A1 exons can be expected as 1/100 (0.1–6.2%, P=0.05) and 0/100 (0.0–4.6%, P=0.05), respectively.
Identification and characterization of breakpoints
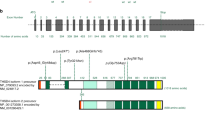
Loss of heterozygosity and decreased signal intensities upon high-density microarray analyses confirmed the MLPA result of patient 53B and narrowed down the deletion breakpoints between rs16829183 and rs932169 (Figure 1b). Accordingly, primers were designed and used for long-range PCR. Because of the distance between the primer-binding sites on the normal allele (∼3.41 Mb), only the amplification of the deletion-carrying allele was possible and resulted in an amplicon of ∼8.5 kb (data not shown). Subsequent sequencing of this amplicon identified a deletion of 3 408 306 bp at 2q32.1q32.3 (Figure 1).

Deletion of 3.4 Mb identified in this study. (a) Schematic representation of the completely deleted genes. Genes are represented by arrows that indicate the direction of transcription according to Entrez Gene (www.ncbi.nlm.nih.gov/sites/entrez?db=gene; version May 2009). COL3A1 is denoted by a black arrow and other genes associated with a dominantly inherited disease and thus considered for further analyses are indicated by gray arrows. Known copy number variations (CNVs) in this region are given below the line according to the Database of Genomic Variants (DGV, http://projects.tcag.ca/variation, version May 2009; Supplementary Table S3). The black bars represent losses (L) and white bars gains (G). (b) Schematic representation of the region flanking the breakpoints. Exons are specified as bars and marked with the corresponding number. Regions derived from high-density microarray analyses are represented by a white bar for loss of heterozygosity and by a blue bar for decreased SNP array signal intensity. The positions of SNPs tested by the array set are indicated by vertical lines. The deleted region is denoted as a brown bar and the primers used for long-range (LR) PCR (LR primers P53B_7F and P53B_17R) are indicated by arrows. (c) Sequence of the long-range PCR product spanning the breakpoint junction of the deletion. Uppercase letters represent the sequence in the region of the start point of the deletion and lowercase letters the sequence in the region of the deletion end point. Because of identical sequences at the site of the breakpoints, the break and rejoining could have occurred at three positions as indicated by open triangles. The dotted line marks the most telomeric position of the possible breakpoints. All nucleotide positions are given in relation to the human genome reference sequence (NCBI build 36.1, March 2006).
At the start and end points of the deletion, there is a short stretch of identical sequences (Figure 1c). This phenomenon has also been reported for other deletions.16, 19 One could speculate that the deletion presented here can be the result of mechanisms mainly responsible for copy number variations (CNVs) in the human genome, such as nonallelic homologous recombination (NAHR), non-homologous end joining (NHEJ), and Fork Stalling and Template Switching (FoSTeS).20
The deletion includes regions with known CNVs (Figure 1a and Supplementary Table S3). Although to date little is known about the conservation and effect of CNVs, the CNVs within the deleted region have been found in healthy individuals and thus considered to be non-pathogenic. This interpretation is also supported by the fact that all known CNVs in the deleted region are relatively small (<200 kb, cf. pathogenic CNVs are mostly >500 kb).21 As expected, microarray analyses of patient 53B also revealed known CNVs in other parts of his genome (data not shown).
Furthermore, the deletion presented here affects not only COL3A1 but also 21 other known genes (Figure 1a). Although the function of most of these genes is unknown/unclear, mutations in three genes (COL5A2, SLC40A1, and MSTN) have previously been associated with autosomal dominant disorders. Accordingly, mutations in COL5A2 (MIM *120190), which encodes the α2 chain of type V collagen, lead to the classical type of EDS (EDS I/II, MIM #130000/#130010), the major diagnostic criteria of which include skin hyperextensibility, widened atrophic scars, and joint hypermobility but no AD.14, 22 The SLC40A1 gene (SLC11A3, MIM *604653) encodes ferroportin and has been associated with hemochromatosis type 4 (HFE4, MIM #606069), a disorder of iron homeostasis.23, 24 MSTN (MIM +601788) encodes the muscle growth inhibitor myostatin and has been found to be mutated in incomplete autosomal dominant muscle hypertrophy (MIM +601788).25 We considered these three genes for further analyses in addition to COL3A1.
Clinical and biochemical phenotypes
Most members of family 53 inherited the deleted allele (Figure 2a). Data on the clinical phenotype of the index patient 53B (Figure 2a) were collected from medical records. Accordingly, 53B died unexpectedly at the age of 34 years because of an abdominal aortic dissection found to be cranial from the celiac artery encompassing all segments of the descending thoracic aorta (Table 1). He was described as being in good general health but with prominent and early-onset varicose veins. In addition to patient 53B, his oldest brother (53, Figure 2a), who was also affected, was the only family member with reported aortic dissection, which occurred at the age of 43, 48, and 51 years, the latter leading to death (Table 1). Autopsy of patient 53 revealed not only the rupture of the thoracic aorta but also the involvement of medium-sized arteries.

Pedigree and some clinical features of Family 53. (a) Pedigree: the arrow denotes the male index patient (53B). The vertical line in the symbols (circle, female; square, male) denotes molecular genetic testing for the 3.4-Mb deletion: white halves represent normal alleles and black ones the allele carrying the deletion. The diagonal line through a symbol indicates deceased family members. The year of birth is given in parentheses. (b) Overfolded ear helix (53I). (c) Small earlobe (53). (d) Prominent varicose veins (53). (e) Muscle hypertrophy of the lower leg (53E).
In contrast, six familial carriers of the 3.4-Mb deletion (53, 53D, 53E, 53H, 53I, and 53J) and one non-carrier (53F), who served as an intrafamilial control subject, were available for physical examinations, which were focused on EDS IV, EDS I/II, muscle hypertrophy, and HFE4 (Figure 2, Table 1, and Supplementary Table S2). From the clinical features of EDS IV, all but one of the deletion carriers showed fragile and thin/translucent skin (Table 1). Varicose veins (Figure 2d) were detected only in adults, whereas hypermobility of small joints was predominantly present in younger deletion carriers. Facial acrogeria was limited to 53 and 53H, whereas acrogeria of hands and feet was present in all but 53I and 53J. Moderately thin lips were also observed (53E, 53I, and 53J). Examination for major clinical signs of EDS I/II revealed smooth velvet skin and joint hypermobility, but none of the other classical clinical signs of EDS I/II were observed (Table 1). Impressively, all investigated male deletion carriers showed increase in muscle size of lower extremities with slightly increased muscle power, a finding consistent with myostatin-related muscle hypertrophy (Figure 2e and Table 1).
Apart from severe (early onset) varicosis and thin/translucent skin, the oldest deletion carrier (53D) is asymptomatic (Table 1). This could be explained by modifying gene(s)/factor(s) or by a mosaic of normal and mutated cells. The latter cannot be excluded, even if analysis of her leukocyte, saliva, and fibroblast DNAs provided no evidence for mosaicism (data not shown).
In addition, standard biochemical blood parameters determined for four deletion carriers (53D, 53E, 53J, and 53I) were comparable with normal values for age and sex (Supplementary Table S2), providing no evidence for HFE4. Notably, although HFE4 has previously been associated with heterozygous mutations in the SLC40A1 gene,23, 24 in these four deletion carriers (53D, 53E, 53J, and 53I) without long-term low iron intake we found neither an increase in serum ferritin nor an abnormal transferrin saturation. In the 15-year-old female deletion carrier (53I), the relative number of hypochrome erythrocytes was slightly increased, but she did not have anemia and all other erythrocyte parameters were within normal range.
Taken together, despite (age related) clinical variability, deletion carriers showed pronounced clinical signs of EDS IV and muscle hypertrophy, but only moderate expression of EDS I/II, resulting in a mixed clinical phenotype of these disorders (Table 1). No other clinical signs and symptoms were observed and no evidence of other disease (eg, cancer) by history was found in deletion carriers.
Biochemical collagen analysis and electron microscopy
In deletion carriers 53, 53B, 53D, and 53E, we detected on SDS-PAGE a normal distribution as well as normal electrophoretic migration patterns for collagens I, III, and V in both medium and cell layer, confirming the normal function of the non-deleted COL3A1 and COL5A2 alleles (patient 53 in Figure 3a). In addition, our biochemical (protein based) collagen analyses confirmed the previous observation that such in vitro testing is less sensitive in identifying mutations that decrease production but do not alter the structure of type III and V collagens (Figure 3a).26, 27

Results of biochemical collagen analysis and electron microscopy. (a) SDS-PAGE of collagens produced by cultured dermal fibroblasts showing normal distribution and electrophoretic mobility of collagen type I (α1(I) and α2(I) chains), collagen type III (homotrimers of α1(III) chains), and collagen type V (α1(V) and α2(V) chains) in patient 53 (P53) and controls (C1, C2, and C3). Note that the α1(V) and α2(V) chains can only be seen on gels exposed for longer time. (b, c) Cross-sections of the dermis of patients 53 (P53) and 53E (P53E) showing abnormal collagen fibrils with either enlarged, irregular outlines (black arrows) or with abnormally small diameters (white arrowheads), occurring within otherwise regularly shaped fibrils. The insert in (b) shows a normal control.
Electron micrographs of the dermis of patients 53, 53D, and 53E showed collagen fibrils with abnormally large diameters and slightly irregular outlines as well as abnormally small collagen fibril diameters, which were interspersed with normal-appearing fibrils (Figures 3b and c). The size variability of fibril diameters was increased compared with the control (cf. insert in Figure 3b). Longitudinal sections of fibrils consisted mostly of normally aggregated filaments as well as of few poorly aggregated ones lacking the typical transversal periodicity (data not shown). All these findings are consistent with a mixed phenotype of EDS IV and EDS I/II.
Discussion
We assessed the contribution of large COL3A1 deletions/duplications to AD in patients in whom genetic testing of FBN1, TGFBR1, and TGFBR2 revealed no mutation. We identified a hemizygous deletion of 3.4 Mb affecting COL3A1 and other genes. Hence, we analyzed the clinical and biochemical effects of the hemizygous deletion of four of these genes (COL3A1, COL5A2, MSTN, and SLC40A1), each of which has previously been associated with an autosomal dominant disorder. Our data show that the hemizygous deletion of COL3A1, COL5A2, and MSTN, but not that of SLC40A1, leads to a clinical phenotype, extending the molecular etiology of EDS IV, EDS I/II, muscle hypertrophy, and HFE4, respectively.
In autosomal dominant disorders, haploinsufficiency can occur when a gene has only a single functional copy, instead of two copies. A distinction is drawn between true and functional haploinsufficiency. True haploinsufficiency is the a priori result of a hemizygous deletion, whereas functional haploinsufficiency occurs when one allele loses functionality, for example, because a key residue is mutated or the transcript amount of the mutant allele is reduced by NMD. As even in the era of CNVs, reports on true haploinsufficiency are rare, functional haploinsufficiency has often been used to assess whether or not haploinsufficiency of a gene is sufficient to cause features of the underlying disease.
A priori, the hemizygous deletion identified in this study causes true haploinsufficiency of the deleted genes (cf. no transcription of the deleted allele). The only question is which of these true haploinsufficiencies is responsible for the clinical phenotype. Thus, one would expect reduced (50%) expression of the affected genes. Indeed, our preliminary quantitative transcript analyses indicated reduction of COL3A1 and COL5A2 transcripts in cultured fibroblasts of deletion carriers (data not shown). However, cell culture conditions as well as modifying gene(s)/factor(s) can influence the expression of the undeleted allele, which can lead to expression levels significantly differing from 50%.28 Furthermore, there is increasing evidence that CNVs can have intra- and inter-chromosomal effects on other genes because of interactions between chromosomal regions.29 Thus, it is possible that the 3.4-Mb deletion identified in this study affects not only the expression of the deleted genes but also other genes on chromosome 2 and/or elsewhere in the genome. Similarly, modifying genetic, epigenetic, environmental, and stochastic factors can be the reason for the (age related) variability in the clinical phenotype of Family 53 (Table 1). For these reasons, we performed physical and biochemical examinations rather than in vitro transcript analyses of the deleted genes.
In previous studies, most of the identified COL3A1 mutations causing EDS IV were missense or splice site mutations, leading to structural alterations of the protein. Only few cases of functional COL3A1 haploinsufficiency, due to NMD, have been reported.27, 30 However, it has remained unclear whether or not the potent dominant-negative effect of the remaining mutant transcripts, which escape NMD, leads to the disease (cf. NMD is almost always incomplete). A recent study on homozygosity for a COL3A1 null allele with NMD (p.Lys161GlnfsX45) could also not resolve this dilemma.30 Cytogenetically detectable 2q32 deletions affect too many genes in addition to COL3A1, leading to severe clinical phenotypes with developmental delay and thus hampering the clinical identification of EDS IV (Supplementary Figure S1 and Supplementary Table S1). In animal models, mice homozygous for an inactivated allele of Col3a1 showed a phenotype closely resembling EDS IV, whereas heterozygous mice were phenotypically normal.31 Late-onset signs in heterozygous mice, however, could not be excluded, because these mice had a limited follow-up period (cf. there are functional changes in bladder tissue of 8-week-old mice heterozygous for a Col3a1 null allele).32 Thus, this study is the first to show at the molecular level that the complete loss of one COL3A1 allele (true haploinsufficiency) can cause an EDS-IV-related phenotype.
Similarly, no cases of true haploinsufficiency of COL5A2 and MSTN have previously been described at the molecular level for EDS I/II and muscle hypertrophy, respectively, the clinical signs of which were clearly shown in this study. In comparison, both true and functional haploinsufficiencies of COL5A1 (MIM *120215), the other gene associated with EDS I/II coding for the α1 chain in the [α1(V)]2, α2(V) heterotrimers of type V collagen, have been reported.33, 34, 35, 36 This difference in the haploinsufficiencies of COL5A2 and COL5A1 may be because of the notion/evidence that homotrimeric formation of α2(V) chains is not possible, whereas α1(V) chains can assemble into stable homotrimers (ie, COL5A1 can replace COL5A2).37, 38 This may have hampered the identification of cases with COL5A2 haploinsufficiency because of lack of sufficient clinical signs. Although AD may occur in EDS I/II,39 none of the few (∼10) COL5A2 mutations reported so far have been associated with AD. In addition, mice with homozygous Col5a2 deletion survived poorly, possibly because of complications from spinal (but not from cardiovascular) deformities, and showed skin and eye abnormalities as a result of disorganized type I collagen fibrils.40 Consequently, the AD phenotype in Family 53 is most likely caused mainly by true haploinsufficiency of COL3A1; the haploinsufficiency of COL5A2 may have merely a modifier effect.
In contrast, we found no evidence for HFE4 in four cases of true SLC40A1 haploinsufficiency, not even in the 77-year-old carrier (cf. normal blood parameters; no signs for joint pains, osteoarthritis, fatigue, cardiomyopathies, and endocrine disorders). This lack of evidence for HFE4 is somewhat unexpected, as heterozygous loss-of-function SLC40A1 mutations have previously been associated with HFE4.41, 42 However, as the age of onset of HFE4 is up to 60 years in males and ∼10 years later in females (age-related penetrance),43 it is possible that young carriers of the SLC40A1 deletion failed to present clinical symptoms of HFE4 in this study because of their age of 15 (53I, female), 18 (53J, male), and 49 (53E, male) years at examination. As, to our knowledge, no patient with complete hemizygous deletion of this gene has previously been reported at the molecular level, this is the first description of the (so far normal) clinical phenotype of individuals with true SLC40A1 haploinsufficiency, suggesting the negligible role of haploinsufficiency in the pathogenesis of HFE4.
Altogether, the hemizygous deletion presented here causes a mixed phenotype because of the true haploinsufficiencies of COL3A1, COL5A2, and MSTN, whereby most likely the true haploinsufficiency of COL3A1 causes AD. In the case of SLC40A1, we provided evidence that a haploinsufficient gene known to be associated with an autosomal dominant disorder does not necessarily lead to a clinical phenotype. Deleted genes that were not further investigated in this study have been associated with a recessive disease or the gene function, and/or their disease association is unknown/unclear. As no evidence of other clinical signs, symptoms, and diseases (eg, cancer) was found in this study, the effect of the haploinsufficiency of these other genes located within the deletion (eg, PMS1) is low, if any. Our finding that true haploinsufficiency leads to an EDS-IV-related phenotype opens the possibility of novel therapeutic strategies, which increase expression of the undeleted allele(s). In addition, this work emphasizes the inclusion of deletion/duplication screening in the comprehensive genetic testing of COL3A1 as well as the importance of this testing in AD patients, at least with EDS-like phenotypes.
References
Pyeritz RE : The Marfan syndrome. Annu Rev Med 2000; 51: 481–510.
De Paepe A, Devereux RB, Dietz HC, Hennekam RCM, Pyeritz RE : Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996; 62: 417–426.
Dietz HC, Cutting GR, Pyeritz RE et al: Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991; 352: 337–339.
Mizuguchi T, Collod-Beroud G, Akiyama T et al: Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 2004; 36: 855–860.
Loeys BL, Chen J, Neptune ER et al: A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275–281.
Pannu H, Fadulu VT, Chang J et al: Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation 2005; 112: 513–520.
Loeys BL, Schwarze U, Holm T et al: Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355: 788–798.
Matyas G, Arnold E, Carrel T et al: Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum Mutat 2006; 27: 760–769.
Germain DP : Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis 2007; 2: 32.
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ : Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 1998; 77: 31–37.
Pope FM, Martin GR, Lichtenstein JR et al: Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc Natl Acad Sci USA 1975; 72: 1314–1316.
Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B : Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem 1988; 263: 6226–6232.
Tromp G, Kuivaniemi H, Shikata H, Prockop DJ : A single base mutation that substitutes serine for glycine 790 of the alpha 1 (III) chain of type III procollagen exposes an arginine and causes Ehlers-Danlos syndrome IV. J Biol Chem 1989; 264: 1349–1352.
Steinmann B, Royce PM, Superti-Furga A : The Ehlers-Danlos syndrome. in: Royce PM, Steinmann B (eds): Connective Tissue and its Heritable Disorders. New York: Wiley-Liss, 2002; 431–523.
Matyas G, De Paepe A, Halliday D, Boileau C, Pals G, Steinmann B : Evaluation and application of denaturing HPLC for mutation detection in Marfan syndrome: identification of 20 novel mutations and two novel polymorphisms in the FBN1 gene. Hum Mutat 2002; 19: 443–456.
Matyas G, Alonso S, Patrignani A et al: Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum Genet 2007; 122: 23–32.
Steinmann B, Rao VH, Vogel A, Bruckner P, Gitzelmann R, Byers PH : Cysteine in the triple-helical domain of one allelic product of the alpha 1(I) gene of type I collagen produces a lethal form of osteogenesis imperfecta. J Biol Chem 1984; 259: 11129–11138.
Vogel A, Holbrook KA, Steinmann B, Gitzelmann R, Byers PH : Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab Invest 1979; 40: 201–206.
Giacalone JP, Francke U : Common sequence motifs at the rearrangement sites of a constitutional X/autosome translocation and associated deletion. Am J Hum Genet 1992; 50: 725–741.
Gu W, Zhang F, Lupski JR : Mechanisms for human genomic rearrangements. Pathogenetics 2008; 1: 4.
Bruno DL, Ganesamoorthy D, Schoumans J et al: Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J Med Genet 2009; 46: 123–131.
Michalickova K, Susic M, Willing MC, Wenstrup RJ, Cole WG : Mutations of the alpha2(V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet 1998; 7: 249–255.
Montosi G, Donovan A, Totaro A et al: Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001; 108: 619–623.
Njajou OT, Vaessen N, Joosse M et al: A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet 2001; 28: 213–214.
Schuelke M, Wagner KR, Stolz LE et al: Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 2004; 350: 2682–2688.
Pepin M, Schwarze U, Superti-Furga A, Byers PH : Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680.
Schwarze U, Schievink WI, Petty E et al: Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet 2001; 69: 989–1001.
Hutchinson S, Furger A, Halliday D et al: Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet 2003; 12: 2269–2276.
Henrichsen CN, Vinckenbosch N, Zollner S et al: Segmental copy number variation shapes tissue transcriptomes. Nat Genet 2009; 41: 424–429.
Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M, Van Kien PK : Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur J Hum Genet 2009; 17: 1411–1416.
Liu X, Wu H, Byrne M, Krane S, Jaenisch R : Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA 1997; 94: 1852–1856.
Stevenson K, Kucich U, Whitbeck C, Levin RM, Howard PS : Functional changes in bladder tissue from type III collagen-deficient mice. Mol Cell Biochem 2006; 283: 107–114.
Toriello HV, Glover TW, Takahara K et al: A translocation interrupts the COL5A1 gene in a patient with Ehlers-Danlos syndrome and hypomelanosis of Ito. Nat Genet 1996; 13: 361–365.
Wenstrup RJ, Florer JB, Willing MC et al: COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am J Hum Genet 2000; 66: 1766–1776.
Bouma P, Cabral WA, Cole WG, Marini JC : COL5A1 exon 14 splice acceptor mutation causes a functional null allele, haploinsufficiency of alpha 1(V) and abnormal heterotypic interstitial fibrils in Ehlers-Danlos syndrome II. J Biol Chem 2001; 276: 13356–13364.
Mitchell AL, Schwarze U, Jennings JF, Byers PH : Molecular mechanisms of classical Ehlers-Danlos syndrome (EDS). Hum Mutat 2009; 30: 995–1002.
Fichard A, Tillet E, Delacoux F, Garrone R, Ruggiero F : Human recombinant alpha1(V) collagen chain. Homotrimeric assembly and subsequent processing. J Biol Chem 1997; 272: 30083–30087.
Chanut-Delalande H, Bonod-Bidaud C, Cogne S et al: Development of a functional skin matrix requires deposition of collagen V heterotrimers. Mol Cell Biol 2004; 24: 6049–6057.
Wenstrup RJ, Meyer RA, Lyle JS et al: Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med 2002; 4: 112–117.
Andrikopoulos K, Liu X, Keene DR, Jaenisch R, Ramirez F : Targeted mutation in the col5a2 gene reveals a regulatory role for type V collagen during matrix assembly. Nat Genet 1995; 9: 31–36.
Wallace DF, Pedersen P, Dixon JL et al: Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood 2002; 100: 692–694.
Schimanski LM, Drakesmith H, Merryweather-Clarke AT et al: In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 2005; 105: 4096–4102.
Cremonesi L, Forni GL, Soriani N et al: Genetic and clinical heterogeneity of ferroportin disease. Br J Haematol 2005; 131: 663–670.
Acknowledgements
We are grateful to Family 53, all patients, and referring physicians for participating in this study. We thank Matthias Baumgartner, Wolfgang Berger, and Albert Schinzel for their support; Philippe Reuge for help with MLPA analyses; Angelika Schwarze for help with cell cultures and biochemical collagen analyses; and Michal Okoniewski, Elisabeth Probst, and members of the Institute of Medical Genetics, University of Zurich, for discussions. This work was supported by the FGCZ and grants from the Swiss National Science Foundation (3100A0–120504 to GM and 3200B0–109370/1 to BS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Meienberg, J., Rohrbach, M., Neuenschwander, S. et al. Hemizygous deletion of COL3A1, COL5A2, and MSTN causes a complex phenotype with aortic dissection: a lesson for and from true haploinsufficiency. Eur J Hum Genet 18, 1315–1321 (2010). https://doi.org/10.1038/ejhg.2010.105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.105
Keywords
This article is cited by
-
Optimising the mutation screening strategy in Marfan syndrome and identifying genotypes with more severe aortic involvement
Orphanet Journal of Rare Diseases (2020)
-
Molecular cytogenetic identification of small supernumerary marker chromosomes using chromosome microarray analysis
Molecular Cytogenetics (2019)
-
Identification of Novel Clinically Relevant Variants in 70 Southern Chinese patients with Thoracic Aortic Aneurysm and Dissection by Next-generation Sequencing
Scientific Reports (2017)
-
Analysis of a gene co-expression network establishes robust association between Col5a2 and ischemic heart disease
BMC Medical Genomics (2013)
