
Abstract
CHARGE syndrome is a multiple congenital anomaly syndrome characterised by Coloboma, Heart defects, Atresia of choanae, Retardation of growth and/or development, Genital hypoplasia, and Ear anomalies often associated with deafness. It is caused by heterozygous mutations in the CHD7 gene and shows a highly variable phenotype. Anosmia and hypogonadotropic hypogonadism occur in the majority of the CHARGE patients, but the underlying pathogenesis is unknown. Therefore, we studied the ability to smell and aspects of the reproductive system (reproductive performance, gonadotropin-releasing hormone (GnRH) neurons and anatomy of testes and uteri) in a mouse model for CHARGE syndrome, the whirligig mouse (Chd7Whi/+). We showed that Chromodomain Helicase DNA-binding protein 7 (Chd7) is expressed in brain areas involved in olfaction and reproduction during embryonic development. We observed poorer performance in the smell test in adult Chd7Whi/+ mice, secondary either to olfactory dysfunction or to balance disturbances. Olfactory bulb and reproductive organ abnormalities were observed in a proportion of Chd7Whi/+ mice. Hypothalamic GnRH neurons were slightly reduced in Chd7Whi/+ females and reproductive performance was slightly less in Chd7Whi/+ mice. This study shows that the penetrance of anosmia and hypogonadotropic hypogonadism is lower in Chd7Whi/+ mice than in CHARGE patients. Interestingly, many phenotypic features of the Chd7 mutation showed incomplete penetrance in our model mice, despite the use of inbred, genetically identical mice. This supports the theory that the extreme variability of the CHARGE phenotype in both humans and mice might be attributed to variations in the fetal microenvironment or to purely stochastic events.
Similar content being viewed by others
Introduction
CHARGE syndrome is a multiple congenital anomaly syndrome with variable occurrence of Coloboma, Heart defects, Atresia of choanae, Retardation of growth or development, Genital hypoplasia, and Ear abnormalities and deafness.1 More recently, semicircular canal hypoplasia and anosmia due to olfactory bulb aplasia were found to be part of the CHARGE spectrum.2, 3, 4, 5 Since the discovery that heterozygous mutations in the CHD7 gene (OMIM #608892) are the major cause of the CHARGE syndrome, the phenotypic spectrum has broadened.6, 7 Some very mild cases of CHARGE syndrome have been identified, some of which were originally diagnosed as Kallmann syndrome.8 A recent study claimed that Kallmann syndrome might be a mild allelic variant of CHARGE syndrome.9 However, it might also be that CHD7 mutations can have a very mild presentation mimicking Kallmann syndrome.8 Kallmann syndrome and CHARGE syndrome share many features including not only anosmia and hypogonadotropic hypogonadism (owing to gonadotropin-releasing hormone (GnRH) deficiency) but also hearing impairment, cleft lip/palate and renal agenesis.8 The underlying cause of this phenotypic overlap is uncertain. It might be that Chromodomain Helicase DNA-binding protein 7 (CHD7), a putative chromatin remodelling protein, could affect the expression of one or several Kallmann syndrome genes (eg, KAL1, FGFR1, PROK2, PROKR2 and FGF8).10 If suitable mouse models are available, mouse studies can provide insights into the underlying pathogenic pathways of anosmia and hypogonadotropic hypogonadism in CHARGE syndrome.
The whirligig mouse (Chd7Whi/+) carries an ENU (N-ethyl-N-nitrosourea)-induced nonsense mutation in the Chd7 gene and shows a phenotype that is very similar to that of humans with CHARGE syndrome.11 Features that are present in both mouse and human include fully penetrant semicircular canal defects and occasional heart defects, choanal atresia, cleft palate and eye defects.11 However, olfaction and reproductive organs have not been extensively studied so far.
We performed a smell test and analysed olfactory bulb and reproductive organ anatomy in Chd7Whi/+ mice. We also studied GnRH neurons in the hypothalamus of embryonic and adult Chd7Whi/+ mice and analysed the expression of Chd7 during olfactory and GnRH neuron development. The overlap between CHARGE and Kallmann syndromes was further explored by crossing Chd7Whi/+ mice with Fgfr1Hspy/+ mice. Fgfr1Hspy/+ mice have a missense mutation (W691R) in the conserved kinase domain of the Fgfr1 gene (one of the genes involved in Kallmann syndrome).12 This missense mutation causes a loss of receptor function (J Calvert, S Dedos, K Hawker and KP Steel, manuscript in preparation).
Methods
Mice and genotyping
Mutant mice were maintained as heterozygote by wild-type matings on a C3HeB/FeJ background.11, 13, 14 Additional matings were set up between Fgfr1Hspy/+ females and Chd7Whi/+ males to create double heterozygous (Hspy/+;Whi/+) mice. The mouse housing and experiments complied with UK Home Office requirements.
Genomic DNA from ear clips was purified15 and genotyping of Fgfr1Hspy/+ (Supplementary Methods) and Chd7Whi/+ mice was performed in submission.11 Wild-type littermates were used as controls.
Olfaction test
In the olfaction tests, we determined the ability of wild-type C3HeB/FeJ mice to discriminate between urine from various mouse strains (adapted from Brown et al16 and Lee et al17). Urine samples from five adult BALB/c and five adult 129/S5 male mice was pooled, diluted with distilled water ((10−2), (10−3) and (10−4)), aliquoted and frozen at −80°C until use. C3HeB/FeJ mice (eight males and five females) aged between 8 and 12 weeks were used for the habituation–dishabituation experiment. Clean plastic mouse cages (∼31 × 12 × 13 cm3) with a gridded lid were placed in a separate room away from the colony. The experiments were conducted in normal light, as C3HeB/FeJ mice are blind because of retinal degeneration from age 3 weeks onwards.18 On day 1, the mice were allowed to become familiar with the test situation by placing them in a clean cage with bedding for 3 min, followed by a 2-min presentation of water (100 μl) on a cotton bud inserted through the lid. On day 2, seven mice were presented with 100 μl water, 3 × 100 μl of 129/S5 urine (10−4) and 1 × 100 μl of BALB/c urine (10−4) on cotton buds for 2 min each, with 2 min between each presentation. The other six mice received 2-min sessions of 100 μl water, 3 × 100 μl of BALB/c urine (10−4) and 1 × 100 μl of 129/S5 urine (10−4). On day 3, all urine concentrations were raised to (10−3) and on day 4 to (10−2). During each 2-min session, the number of sniff bouts and their cumulative duration were recorded.
For the olfaction test, we used 21 Chd7Whi/+ mice aged between 11 and 15 weeks (9 females and 12 males), and 19 age- and sex-matched wild-type littermates (8 females and 11 males). On day 1, all mice became familiar with the test situation as described above. On day 2, all mice were presented with 100 μl water followed by 100 μl of BALB/c urine (10−2) for 2 min each, with 2 min between the presentations. During each 2-min session, the number of sniff bouts and their cumulative duration were recorded. The data of Chd7Whi/+ and wild-type mice were compared using the Wilcoxon signed-rank test (as in Lee et al17).
Histopathological examination
Mice used for the smell test (n=40) were weighed, injected with pentobarbital sodium BP (Lethobarb, Fort Dodge Animal Health, Southampton, UK) and transcardially perfused with ice-cold 10% formalin (Sigma, Gillingham, UK). Brain, testes, ovaries and uteri were dissected, weighed and preserved in 10% neutral buffered formalin (Sigma). Gross anatomy was viewed using a Leica stereomicroscope and digital photographs were taken with a Leica Dfc490 camera. The olfactory bulb and brain lengths, used to establish the olfactory bulb/brain ratio, were measured as depicted in Figure 3a. Testes were weighed and their length was measured. Uterine horns and ovaries were weighed together. Measurements of the Chd7Whi/+ and wild-type brains and reproductive organs were analysed using Student's t-test (or the Mann–Whitney test for small sample sizes, n<15). Brains were processed for paraffin embedding and cut in 8 μm coronal sections with every 10th section mounted on a SuperFrost (VWR, Lutterworth, UK) glass slide. Olfactory bulb organisation was analysed in Nissl-stained sections.19
E12.5 and E16.5 wild-type, Chd7Whi/+ and Hspy/+;Whi/+ embryos were obtained from timed pregnancies, with E0.5 at noon on the day the vaginal plug was found. The embryos (n=28) were dissected in ice-cold PBS and yolk sacs were taken for genotyping.11 Heads were fixed in 10% neutral-buffered formalin, dehydrated and embedded in paraffin. Sections of 8 μm thickness were stained for haematoxylin/eosin or processed for immunohistochemistry, for which the Ventana Discovery System (Ventana, Tucson, AZ, USA) and Ventana reagents (Ezprep, cat. no. 950-100), CC1 (cat. no. 950-124), LCS (cat. no. 50-010), Reaction buffer (cat. no. 950-300), DABMap Kit (cat. no. 760-124) and haematoxylin counterstain (cat. no. 760-2021) were used according to the manufacturer's instructions. Antibodies against GnRH1 (Chemicon, Watford, UK, cat. no. MAB5456) and CHD7 (a gift from Abcam, Cambridge, UK, cat. no. ab31824) were diluted 1:500 in Ventana buffer (cat. no. 251-018, Ventana). The respective secondary antibodies, biotinylated rabbit anti-mouse and biotinylated donkey anti-rabbit, were diluted 1:100 in PBS containing 10% heat-inactivated fetal calf serum, 2% BSA, 0.1% Triton X-100 and 10 mM sodium azide. The sections were examined with a Zeiss Axioscope (Welwyn Garden City, UK) and digital images were taken using a Zeiss Axioscope HRc.
Breeding data analysis
The reproductive function of adult wild-type (mean age 2.2 months, SD 0.77) and Chd7Whi/+ (mean age 3.2 months, SD 1.76) mice was assessed by the number of days required for production of the first litter after placing the test mutant with a wild-type mate. Differences were analysed with Student's t-test (or the Mann–Whitney test for small samples).
Results
Chd7 is present in brain areas involved in olfaction and reproduction
Immunohistochemistry tests in wild-type embryos showed decreasing amounts of Chd7 in the brain with increasing age (Figure 1). At E12.5 and E14.5, Chd7 protein was present in all layers of the olfactory epithelium and olfactory bulb (Figure 1a and b). At E16.5, the expression became more restricted, but Chd7 was detected in the vomeronasal organ, olfactory epithelium, olfactory bulb, hypothalamus and pituitary (Figure 1c–f). In the olfactory epithelium, Chd7 expression was highest in the inner basal layer, which contains horizontal and globose basal cells. Chd7 expression was also present in the nuclei of olfactory sensory neurons in the intermediate layer. The apical layer with supportive sustentacular cells showed the lowest expression of the Chd7 protein (Figure 1d).

Immunohistochemistry showed that Chd7 protein is present in olfactory epithelium and in areas of the brain involved in olfaction and reproduction in wild-type C3HeB/FeJ mice. (a) At E12.5 and (b) E14.5, Chd7 is present in the olfactory epithelium and olfactory bulb (sagittal sections). (c–f) At E16.5, Chd7 is present in the vomeronasal organ and olfactory epithelium, olfactory bulb, hypothalamus and pituitary (coronal sections). (d) Chd7 expression is most prominent in the basal cells and olfactory sensory neurons. Scale bars=500 μm (a and b); 100 μm (c, e and f); 50 μm (d). bc, basal cells; hypo, hypothalamus; ns, nasal septum; ob, olfactory bulb; oe, olfactory epithelium; osn, olfactory sensory neurons; pit, pituitary; rp, Rathke's pouch; ssc, supportive sustentacular cells; to, tongue; and vno, vomeronasal organ.
Chd7Whi/+ mice performed worse than wild-type mice on olfaction test
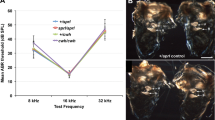
During the habituation–dishabituation test, we found that wild-type C3HeB/FeJ mice showed no interest in 129/S5 urine, but did respond to BALB/c urine from 10−3 concentration onwards (data not shown). Therefore, BALB/c urine at 10−2 concentration was used to assess any difference in smell behaviour between wild-type and Chd7Whi/+ mice. Wild-type mice (n=19) showed an increased response to urine compared with water (P<0.01; Wilcoxon test; Figure 2). Chd7Whi/+ mice (n=21) also showed an increased response to urine compared with water (P<0.05 for the number of sniff bouts and P<0.01 for the cumulative duration of sniff bouts; Figure 2), but the response was less than that seen in wild-type mice (P<0.01; Figure 2). In addition, the number of non-responders, defined as those mice that did not explore the urine,16 was greater in the Chd7Whi/+ mice (5/21) compared with wild-type mice (1/19).

Olfaction test in wild-type and Chd7Whi/+ mice. (a) Mean number of sniff bouts on presentation of water and urine. (b) Mean cumulative duration of sniff bouts. Error bars are means±SEM. *P<0.05.
Chd7Whi/+ mice have mild olfactory bulb hypoplasia
Macroscopic examination of the Chd7Whi/+ and wild-type brains was carried out to analyse the olfactory bulb/brain length ratio (Figure 3a–c and f). Chd7Whi/+ mice (n=23) displayed a slightly decreased olfactory bulb/brain length ratio compared with wild-type controls (n=16) (P<0.01; Student's t-test; Figure 3f). Abnormal olfactory bulbs were seen in two Chd7Whi/+ mice; one olfactory bulb consisted of two parts (Figure 3b) and in one case asymmetrical olfactory bulb hypoplasia was observed (Figure 3c). Microscopic analysis of olfactory bulbs in Chd7Whi/+ mice showed no abnormalities in the layered organisation of the olfactory bulb (Figure 3d and e).

Macroscopic and microscopic analysis of olfactory bulbs from wild-type and Chd7Whi/+ mice. (a–c) Macroscopic view of brains of adult wild-type and Chd7Whi/+ mice, with the olfactory bulb/brain length ratio measured as mean olfactory bulb length divided by length of cerebral hemispheres (arrows in a). (b) Left olfactory bulb consisting of two parts, (c) asymmetrical olfactory bulb hypoplasia. (d, e) Nissl-stained coronal sections of adult wild-type and Chd7Whi/+ olfactory bulbs, showing no abnormalities. (f) Scatter plot of the olfactory bulb/brain length ratio of adult wild-type and Chd7Whi/+ mice. Scale bars=5 mm (a–c); 100 μm (d and e). gl, glomerular cell layer; ml, mitral cell layer; and pgl, periglomerular layer.
Abnormal reproductive system in Chd7Whi/+ mice
Reproductive organs were examined at a macroscopic level (Figure 4). The mean testis weight of adult Chd7Whi/+ (n=12) males was less than for adult wild-type males (n=11) (P≤0.05; Mann–Whitney test; Table 1). However, as Chd7Whi/+ mice had a lower body weight, they had a significantly raised testis weight/body weight ratio (gonadosomatic index, GSI, P<0.05; Mann–Whitney test; Table 1, Figure 4c). Of 12 Chd7Whi/+ males, 2 had severely hypoplastic testes with a decreased GSI (Figure 4b and c). The combined weights of the uteri and ovaries did not differ between wild-type and Chd7Whi/+ mice (Table 1), but abnormalities were present in all Chd7Whi/+ females (n=9) (Figure 4e and f). We identified a cyst in one uterine horn (data not shown) and one unilateral hypoplastic uterine horn (Figure 4e), and all other uteri were shorter and wider than in wild-type mice (Figure 4f).

Macroscopic view of male and female reproductive organs of adult wild-type and Chd7Whi/+ mice. (b) Hypoplastic testes in some Chd7Whi/+ males. (c) Scatter plot of gonadosomatic index of adult wild-type and Chd7Whi/+ males. (e–f) Abnormal uteri in all Chd7Whi/+ females, (e) unilateral hypoplastic uterine horn and (f) wider than normal uterine horns. Scale bars=5 mm (a and b); 1 cm (d–f).
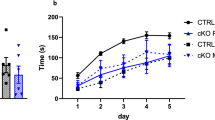
The reproductive performance of Chd7Whi/+ mice was slightly impaired. Maintaining the colony was not a major problem, but the number of days required to produce the first litter was significantly greater in both male and female Chd7Whi/+ mice than in wild-type mice (P<0.05, Mann–Whitney test or t-test, Table 1). The percentage of matings that did not lead to a litter 2 months after set-up was also counted (matings of mutant with wild-type compared with wild-type matings). In wild-type mice, the percentage of matings not producing a litter within 2 months after set-up was 6.1%. The percentage of the Chd7Whi/+ × wild-type matings that did not produce a litter after 2 months was not significantly different from that of wild-type × wild-type matings.
GnRH1 neurons in developing and adult Chd7Whi/+ mice
An immunohistochemistry test with an anti-GnRH1 antibody was carried out to detect GnRH neurons in the adult brain. GnRH1 neurons were present in the organum vasculosum of the lamina terminalis and their fibres extended to the median eminence (ME) of adult wild-type and Chd7Whi/+ mice (Figure 5). Representative photomicrographs were taken to analyse the GnRH neuron density, which appeared to be reduced in the ME of adult Chd7Whi/+ females compared with wild-type females (Figure 5g and h).

Representative photomicrographs of coronal brain sections of adult wild-type and Chd7Whi/+ mice showing GnRH1-positive cells labelled by immunohistochemistry. (a–d) GnRH1 neurons in the organum vasculosum of the lamina terminalis (OVLT). (e–h) GnRH1 axon terminals in the median eminence (ME). Scale bars=100 μm.
No morphological abnormalities were found in the olfactory epithelia or olfactory bulbs of the Chd7Whi/+ embryos at E16.5 (Supplementary Figure 1). GnRH1-positive cells were detected along their migration path, alongside the nasal septum, in the olfactory bulb and in the hypothalamus of the wild-type and Chd7Whi/+ embryos (Supplementary Figure 1a–f). No visible difference in the GnRH1 neuron density between the wild-type and Chd7Whi/+ embryos could be detected (Supplementary Figure 1).
Combining mutations in Fgfr1 and Chd7 is lethal
We collected offspring from matings between Fgfr1Hspy/+ females and Chd7Whi/+ males. No double heterozygous (Hspy/+;Whi/+) animals were recovered at weaning (0/55), showing a significant deviation from normal Mendelian ratios (χ2-test, P<0.01). Subsequently, we collected embryos at E12.5, E16.5 and P0 and found 8 out of 37 Hspy/+;Whi/+ mice, with 2 at P0 (1 dead). This corresponds with the normal Mendelian ratios (χ2-test, P>0.9). This suggests that the combination of both heterozygous mutations results in perinatal or early postnatal death. No anatomical abnormalities were observed in E16.5 Hspy/+;Whi/+ embryos (n=5), other than the anomalies present in Chd7Whi/+ mice with variable penetrance (cleft palate, choanal atresia, heart defect; data not shown). Furthermore, a normal distribution of the GnRH1 neurons was present in E16.5 Hspy/+;Whi/+ embryos (data not shown).
Discussion
The Chd7 protein distribution described here is similar to the Chd7 mRNA distribution previously described5, 9, 11 and corresponds to the β-galactosidase activity in mice carrying a Chd7Gt allele.20 In the olfactory epithelium, the highest levels of Chd7 were found in the basal cells and olfactory sensory neurons, suggesting a role for Chd7 in the development of these cell types. We also detected Chd7 in the developing olfactory bulb and the hypothalamus, pointing to Chd7 having a role in the development of these organs.
Sense of smell was evaluated with urine as an attractive odour, as previous studies had shown that mice are not interested in other odours.17 Urine contains pheromones as well as volatile constituents of urinary odour types and is detected by both the vomeronasal organ and the olfactory epithelium.21, 22, 23 The smell test we used could therefore not distinguish between actions of the vomeronasal organ and the olfactory epithelium. As Chd7 is expressed in both organs, we expect that both could be affected by Chd7 mutations. Overall, Chd7Whi/+ mice performed worse than did wild-type mice, implying that they might have a smell deficit. However, within the mutant group, some mice performed well and seemed to have a normal sense of smell, whereas other mice did not explore the cotton bud dipped in urine (non-responders). The non-responders could be anosmic, but alternatively severe balance disturbances could also have contributed to their poor performance on the smell test. Our study design did not allow discrimination between olfactory and balance/neuromotor dysfunction. However, a concurrent study by Layman et al24 has confirmed the presence of a smell deficit in Chd7-deficient mice (Chd7Gt/+ mice), with an odour-evoked electro-olfactogram.
Pathological examination of the brains of the Chd7Whi/+ mice showed mild hypoplasia of the olfactory bulbs, with only 2 mice (out of 23) having overt olfactory bulb anomalies. The organisation of the layers of the olfactory bulb was normal. These findings are in concordance with a concurrent study.24 One could argue whether these mild olfactory bulb anomalies would have any effect on the ability to smell. It is also possible that a dysfunction of the olfactory epithelium causes the smell deficit, as was postulated by Layman et al.24 The low incidence of olfactory bulb anomalies in Chd7Whi/+ mice are in contrast to the previously described complete penetrance of olfactory bulb aplasia in humans.3, 4, 5 However, we know from our own observations that some patients with a CHD7 mutation have normosmia or slight hyposmia (5/24, JEH Bergman and CMA van Ravenswaaij-Arts, unpublished results).
Defects of reproductive organs were seen in all Chd7Whi/+ females and some Chd7Whi/+ males. Of 12 Chd7Whi/+ males, 2 had severely hypoplastic testes and all 9 Chd7Whi/+ females had abnormal uterine horns. GnRH1 neurons were present in the hypothalamus of Chd7Whi/+ mice, but were mildly decreased in female Chd7Whi/+ mice. In contrast, no GnRH1 neuron abnormalities were observed in Chd7Whi/+ embryos, suggesting that Chd7 might have an effect on the GnRH1 neuron survival in a similar way to Fgfr1.25 The effect of the reproductive organ anomalies and assumed reduction in the GnRH1-positive neurons on reproductive performance seemed minimal, as maintaining the colony did not present any major problems. This is to be expected, as there is a substantial redundancy known to be present in the GnRH neuronal population.26 However, on further analysis, Chd7Whi/+ mice were found to have a slightly lower reproductive performance (needing more days to produce the first litter compared with wild-type mice). Reproductive fitness could be affected by balance defects leading to abnormal head-bobbing and circling behaviour, by reduced body weight or by abnormal mating behaviour of Chd7Whi/+ mice. Reduced body weight was previously correlated with reduced reproductive fitness in mice.27 The cause of reduced body weight in Chd7Whi/+ mice is unknown, but could be caused by increased activity, poor feeding (owing to cranial nerve anomalies or olfactory deficit) or growth hormone deficiency. Mating behaviour was not observed in this study, but it was previously shown that anosmic mice can mate and are fertile.28 However, other studies29, 30 claim that anosmia could lead to reduced reproductive fitness in mice. We were unable to differentiate between the different mechanisms that could influence reproductive fitness because some features that might affect reproductive fitness are fully penetrant in Chd7Whi/+ mice (eg, balance disturbance). The ability of Chd7Whi/+ mice to reproduce corresponds with the situation in a minority of humans with CHARGE syndrome, in which some mildly affected patients are seen to reproduce normally.31, 32
Anosmia and hypogonadotropic hypogonadism often occur together because of the interlinked migration process of olfactory neurons and GnRH neurons during embryonic development.33 However, in our study, poor performance on the smell test did not correlate with hypoplasia of reproductive organs in Chd7Whi/+ mice. Hypoplasia of reproductive organs was also not associated with a clearly reduced GnRH1 neuronal population in the hypothalamus, implying that gonadal anomalies in Chd7Whi/+ mice might be caused by a local effect of Chd7.
The phenotypic overlap between CHARGE and Kallmann syndromes might be attributed to an interaction between the genes (or their encoded proteins) involved in both syndromes. Intercrosses between Fgfr1Hspy/+ females and Chd7Whi/+ males showed that combining mutations in both Fgfr1 and Chd7 leads to perinatal or early postnatal death. This is in contrast to the normal viability of Fgfr1Hspy/+ mice and 50% lethality of Chd7Whi/+ mice, suggesting that the mutations in Fgfr1 and Chd7 interact synergistically, showing more than simply an additive effect on viability. As Chd7 is a member of the CHD family,34, 35 one can hypothesise that reduced levels of Chd7 may decrease Fgfr1 expression, leading to the Kallmann-like phenotype in CHARGE syndrome patients. However, the Kallmann phenotype (olfactory bulb defects and hypogonadotropic hypogonadism) was not observed in double heterozygote mice. This argues against genetic interaction between Fgfr1 and Chd7 in olfactory bulb and hypothalamus development.
Conclusions
Whirligig mice show a reduced penetrance of features associated with anosmia and hypogonadotropic hypogonadism compared to humans with CHARGE syndrome. The observation of incomplete penetrance in mice that have the same Chd7 mutation on an identical genetic background is in line with the variable expression and reduced penetrance observed in humans with CHARGE syndrome. The reduced penetrance and variable expression in these mice may be attributed to purely stochastic events, or may be influenced by environmental differences between mice in their fetal microenvironment. In humans, the modifying effects of the varied genetic backgrounds of people with CHD7 mutations is also likely to influence the penetrance and expression of each feature of the syndrome. Chd7Whi/+ mice may be useful in investigating the potential influence of epigenetic alterations on different aspects of the phenotypic spectrum in these syndromes.
References
Pagon RA, Graham Jr JM, Zonana J, Yong SL : Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 1981; 99: 223–227.
Amiel J, Attiee-Bitach T, Marianowski R et al: Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome. Am J Med Genet 2001; 99: 124–127.
Chalouhi C, Faulcon P, Le Bihan C, Hertz-Pannier L, Bonfils P, Abadie V : Olfactory evaluation in children: application to the CHARGE syndrome. Pediatrics 2005; 116: e81–e88.
Pinto G, Abadie V, Mesnage R et al: CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab 2005; 90: 5621–5626.
Sanlaville D, Etchevers HC, Gonzales M et al: Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet 2006; 43: 211–217.
Vissers LE, van Ravenswaaij CM, Admiraal R et al: Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004; 36: 955–957.
Jongmans MC, Admiraal RJ, van der Donk KP et al: CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 2006; 43: 306–314.
Jongmans MCJ, van Ravenswaaij-Arts C, Pitteloud N et al: CHD7 mutations in patients initially diagnosed with Kallmann Syndrome – the clinical overlap with CHARGE syndrome. Clin Genet 2009; 75: 65–71.
Kim HG, Kurth I, Lan F et al: Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 2008; 83: 511–519.
Hardelin JP, Dode C : The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sex Dev 2008; 2: 181–193.
Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP : Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet 2005; 14: 3463–3476.
Dode C, Fouveaut C, Mortier G et al: Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat 2007; 28: 97–98.
Hawker K, Fuchs H, Angelis MH, Steel KP : Two new mouse mutants with vestibular defects that map to the highly mutable locus on chromosome 4. Int J Audiol 2005; 44: 171–177.
Pau H, Hawker K, Fuchs H, de Angelis MH, Steel KP : Characterization of a new mouse mutant, flouncer, with a balance defect and inner ear malformation. Otol Neurotol 2004; 25: 707–713.
Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A : Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 1999; 126: 1631–1642.
Brown RE, Singh PB, Roser B : The major histocompatibility complex and the chemosensory recognition of individuality in rats. Physiol Behav 1987; 40: 65–73.
Lee AW, Emsley JG, Brown RE, Hagg T : Marked differences in olfactory sensitivity and apparent speed of forebrain neuroblast migration in three inbred strains of mice. Neuroscience 2003; 118: 263–270.
Farber DB, Lolley RN : Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 1974; 186: 449–451.
Kawamura S, Niimi K : Counterstaining of Nauta–Gygax impregnated sections with cresyl violet. Stain Technol 1972; 47: 1–6.
Hurd EA, Capers PL, Blauwkamp MN et al: Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome 2007; 18: 94–104.
Boehm U : The vomeronasal system in mice: from the nose to the hypothalamus and back!. Semin Cell Dev Biol 2006; 17: 471–479.
Kelliher KR : The combined role of the main olfactory and vomeronasal systems in social communication in mammals. Horm Behav 2007; 52: 561–570.
Restrepo D, Arellano J, Oliva AM, Schaefer ML, Lin W : Emerging views on the distinct but related roles of the main and accessory olfactory systems in responsiveness to chemosensory signals in mice. Horm Behav 2004; 46: 247–256.
Layman WS, McEwen DP, Beyer LA et al: Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum Mol Genet 2009; 18: 1909–1923.
Tsai PS, Moenter SM, Postigo HR et al: Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population. Mol Endocrinol 2005; 19: 225–236.
Herbison AE, Porteous R, Pape JR, Mora JM, Hurst PR : Gonadotropin-releasing hormone neuron requirements for puberty, ovulation, and fertility. Endocrinology 2008; 149: 597–604.
Bünger L, Lewis RM, Rothschild MF, Blasco A, Renne U, Simm G : Relationships between quantitative and reproductive fitness traits in animals. Phil Trans R Soc 2005; 360: 1489–1502.
Belluscio L, Gold GH, Nemes A, Axel R : Mice deficient in Golf are anosmic. Neuron 1998; 20: 69–81.
Kelliher KR : The combined role of the main olfactory and vomeronasal systems in social communication in mammals. Horm Behav 2007; 52: 561–570.
Vandenbergh JG : Effects of central and peripheral anosmia on reproduction of female mice. Physiol Behav 1973; 10: 257–261.
Delahaye A, Sznajer Y, Lyonnet S et al: Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clin Genet 2007; 72: 112–121.
Jongmans MC, Hoefsloot LH, van der Donk KP et al: Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet A 2008; 146: 43–50.
Cariboni A, Maggi R : Kallmann's syndrome, a neuronal migration defect. Cell Mol Life Sci 2006; 63: 2512–2526.
Hall JA, Georgel PT : CHD proteins: a diverse family with strong ties. Biochem Cell Biol 2007; 85: 463–476.
Marfella CG, Imbalzano AN : The Chd family of chromatin remodelers. Mutat Res 2007; 618: 30–40.
Acknowledgements
We thank Lianne Stanford for advice on olfaction tests, Jenny Salisbury for help with urine collection, Jeanne Estabel for help with necropsy, Yvette Hooks and Kay Clark for histological stainings, Hermien de Walle for statistical analysis and Jackie Senior and Professor Robert Hofstra for help with preparation of the paper. This work was supported by the Wellcome Trust, the Medical Research Council (UK) and The Netherlands Organisation for Health Research and Development (Grant no. 92003460 to JEHB).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Bergman, J., Bosman, E., van Ravenswaaij-Arts, C. et al. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur J Hum Genet 18, 171–177 (2010). https://doi.org/10.1038/ejhg.2009.158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.158
Keywords
This article is cited by
-
Combined pituitary hormone deficiency harboring CHD7 gene missense mutation without CHARGE syndrome: a case report
BMC Endocrine Disorders (2023)
-
Transcriptional and Epigenetic Control of Mammalian Olfactory Epithelium Development
Molecular Neurobiology (2018)
-
CHD7 promotes proliferation of neural stem cells mediated by MIF
Molecular Brain (2016)
-
A missense mutation in Fgfr1 causes ear and skull defects in hush puppy mice
Mammalian Genome (2011)

{kind=link}