
Abstract
Two intervals of sequence identity at the tips of X and Y chromosomes, the human pseudoautosomal regions PAR1 and PAR2, have drawn interest from researchers in human genetics, cytogenetics, and evolutionary biology. However, they have been widely ignored in linkage and association studies. The pseudoautosomal regions (PARs) pair and recombine during meiosis like autosomes, but the recombination activity in PAR1 is extremely different between sexes. In men, it exhibits the highest recombination frequencies of the genome. Conflicting genetic maps of this region have been estimated by using three-generation pedigrees, sperm typing, and by using haplotypes from single nucleotide polymorphisms. Male genetic map lengths in the literature vary, and linkage disequilibrium has not been analyzed in detail. We review existing tools like genetic and physical maps, linkage disequilibrium methods, linkage and association analysis, implemented statistical methods, and their suitability for PARs. For multipoint linkage analysis, sex specificity must be indicated twice, first using sex-specific maps, and second by considering the sex-specific pseudoautosomal inheritance pattern. Currently, microsatellite panels and single nucleotide polymorphism chips do not contain sufficient numbers of markers in PAR1 and PAR2. The number of markers in PAR1, needed in indirect association studies, should be much larger than for autosomal regions alike in size, since linkage disequilibrium is very low. For genome-wide studies, it is essential to include pseudoautosomal markers since such expensive studies cannot afford to oversee pseudoautosomal linkage or association. This drawback could be solved with a sufficient number of markers, statistical methods that are adopted for the PARs, and their integration into softwares.
Similar content being viewed by others

Introduction
Sex chromosomes have been an issue of interest for decades because of their distinctive pattern of transmission and their peculiar structure and function. The human sex chromosomes, X and Y, are morphologically and genetically distinct. In humans, women have both X chromosomes alike in size and genetic content. Pairing and recombination can exist along their entire length. Men have one X and one Y chromosome and only two limited regions of identical sequence, located at the tips of the short and long arm of the X and Y chromosomes, as depicted in Figure 1. Their physical lengths are approximately 2.7 Mb for PAR1 and 0.33 Mb for PAR2. During meiosis, pairing and crossover in men takes place in these two regions and therefore they have been termed pseudoautosomal regions or ‘PARs’.1, 2 The pseudoautosomal boundaries separate highly recombinant regions from nonrecombinant regions on the Y and a moderately recombining region on the X chromosome.

Structure of the human male sex chromosomes and the localization of the two regions PAR1 and PAR2.
A loss of PAR1 has been observed to be associated with male sterility leading to the theory that the existence of PAR1 is necessary for homologous X–Y chromosome pairing and the proper segregation of gametes.3, 4 This is of interest because the region is physically relatively small. It is estimated that during male meiosis on average at least one crossover occurs. Crossover activity in PAR1 is much higher in men than in women and also higher than for each of the autosomes.5, 6, 7, 8, 9 As a consequence of the elevated recombination rate in this region approximately one half of male children carries a recombinant PAR1 on their Y chromosome whereas the other half inherits a nonrecombinant PAR1 haplotype from their fathers. The rate of recombination in PAR2 is much lower than in PAR1 but still higher than the average rate of the remainder of the X chromosome.
To date, 24 genes have been reported in PAR1 and 5 genes in PAR2.10 Possible connections with clinical disorders such as short stature, asthma, psychiatric disorders, and leukemia have been suggested in the past, but only one pseudoautosomal gene, SHOX (short stature homeobox), has been unambiguously associated with various short stature conditions and disturbed bone development. The SHOX gene has been correlated to disease via deletion mapping and its functions have been recently reviewed.11 However, in systematic genome-wide linkage analysis, PARs have been largely neglected so far – a systematic ‘blind spot’ in genome scans.
This review focuses on statistical methods for genetic map construction, linkage analysis and genetic association analysis in the PARs. It summarizes genetic map estimates, pseudoautosomal markers available on single nucleotide polymorphism (SNP) chips, and methodological developments to account for the special characteristics of the PARs in parametric and nonparametric linkage analysis as well as genetic association analysis.
Brief history
The origin of the human sex chromosomes can be traced in the past by comparing their sequence, gene content, and gene function with related species. The topic has been extensively studied, we refer to some recent reviews.12, 13, 14 It was first postulated in 1914 that X and Y chromosomes evolved from a pair of autosomes and have become separated in the course of hundreds of millions of years.15 The evolution of sex chromosomes emerged when one strand of a pair of autosomes obtained a sex-determining gene. Over time, additional genes with male-specific functions accumulated in this sex-determining chromosome started losing the ability to recombine with its counterpart. Subsequently, a degeneration of size started due to gene inactivation, mutation, deletion, and insertion of junk. It is still an open question why the Y chromosome degenerated so quickly and why positive selection of male advantage genes did not work stronger against it. The theory that the Y could even disappear is maintained by comparative studies in other vertebrates. Aitken and Graves16 predicted a complete loss of the Y chromosome in about 10 million years. In human, Haldane17 found evidence for partial sex linkage in 1936. Later, partial sex linkage was demonstrated to occur in the short arms of X and Y chromosomes by studying spermatocytes in meiotic prophase and during pairing.18, 19 About 60 years after the discovery of PAR1, a second pseudoautosomal region (PAR2) at the opposite ends of the X and Y chromosomes (Xq/Yq) was detected.2
Recombination rate estimation and genetic maps
General considerations
A genetic map is a sequence of genetic loci with distances between adjacent loci reflecting crossover activity. Physical and genetic maps are essential for linkage and association studies. Genetic maps are based on the crossover process. At meiosis, after the duplication of each chromosome, the two pairs of each autosome form a bundle of four strands where chiasmata can occur. For modeling purposes, it is assumed that each chiasma causes two non-sister chromatides to crossover and the pair of strands involved is randomly selected, a process which is called no chromatid interference. A genetic measure of distance is defined as the expected number of crossovers between two loci on a single chromatid (measured in Morgan, M). Since only recombinants (uneven number of crossovers) can be observed, the relation between recombination rate θ and genetic distance x has to be specified by so called mapping functions. In case of complete interference, one chiasma completely suppresses others on the same chromosome, and Morgan's map function20 x=θ is justified. On the other extreme, complete absence of interference leads to the Haldane map function.21 Although in human, the assumption of independent occurrence of crossovers in adjacent intervals holds approximately, finer analysis reveals that the presence of one chiasma reduces the creation of a second one nearby, resulting in positive interference. Moderate interference is assumed by Kosambi's map function.22 For sufficiently long distances (if θ tends to 1/2) the interference vanishes, and for very short segments it increases to 1. It is the most used map function since it seems to adequately reflect the level of interference observed in the human and other mammals. More sophisticated models have been investigated by several authors.23, 24, 25, 26 For example, Sturt developed mapping functions with and without considering chromatid interference under the assumption that at least one crossover occurs on a chromosome arm.23, 24
Recombination rate estimation in PARs
Three different strategies are used to estimate recombination rates for the construction of genetic maps: (1) three-generation families, (2) sperm typing in single men, and (3) unrelated individuals. In sperm typing studies, only male recombination rates can be estimated. In three-generation studies estimations of sex-specific recombination rates are allowed but only generate maps above the megabase scale. Using unrelated individuals, a very fine resolution is reached but only sex-averaged recombination rates can be estimated.
Three-generation families
Genetic maps of three-generation families allow to locate the order of loci as well as estimate sex-averaged and sex-specific recombination rates with linkage analysis, for details we refer to Liu.27 The first genetic map of the human genome was created in 1987 by the Centre d’Etude du Polymorphisme Humaine (CEPH), followed by the Marshfield map,28 and the deCODE map.29 The most recent Rutgers map30 combined genotypes from 14 759 microsatellite and SNP markers on CEPH and deCODE pedigrees. Although this last map is based on 2000 meioses, its resolution is still limited. Generally, the possible resolution depends on the number of analyzed meioses. For example, the estimation of a recombination rate of 0.5% with 95% confidence interval of width 0.25% requires 12 000 informative meioses. Several attempts have been made to integrate multiple types of mapping data. Most recently, Duffy31 used weighted regression to obtain smoothed local recombination rates to interpolate between markers with known genetic distances. Mapping studies that focused on PARs are shown in Table 1.
Single-sperm typing
In sperm typing, alleles present on single haploid cells can be typed and thus haplotypes are determined without pedigree analysis.36 The method allows studying recombination on a fine scale since a large number of meiosis can be analyzed from a single male. Multipoint recombination rate estimation modified to take into account typing errors, contamination rates, and the occurrence of multiple sperm per sample is generally used.37, 32 Two sperm typing studies in PARs have been reported32, 33 and are depicted in Table 1. It has been shown both in bovine and human PARs that recombination exhibits a significant individual variability.33, 38 Therefore, ignoring individual variability of recombination rate estimates including only very few men but many sperms leads to an underestimation of standard errors. In addition, genetic maps from sperm typing studies could be biased since not all sperm represent viable gametes.
Unrelated individuals
Haplotypes from unrelated individuals also bear information on the recombination rates. They reflect linkage disequilibrium (LD) on the population level since LD describes the presence of nonrandom association between two ore more alleles at distinct loci. The LD is reduced if recombination occurs during transmission into the next generation. Since other evolutionary forces influence the LD, recombination rate estimations from haplotypes require some assumptions about the history like population size, population structure, mutation, and selection. Recent statistical methods, based on the coalescence theory, have made it feasible to estimate recombination rates using unrelated individuals.39, 40 They enable a very fine resolution in densely typed regions by taking into account historic recombination events. The results have been compared with those from pedigrees and displayed a strong concordance for the majority of the regions with only some discrepancies at the end of the chromosomes. The information from previous generations is very helpful to estimate recombination rates in regions with low recombination such as PAR2. It has been applied to HapMap SNP genotypes for the human genome at the kilobase scale (phase 2, 2005). Two disadvantages are that (1) a coalescent model of a finite neutrally evolving population with constant population size is assumed which cannot be easily validated with empirical data and (2) only sex-averaged recombination rates can be estimated.
Results for the pseudoautosomal regions
Genetic features of the PARs are summarized in Table 2. Both regions, although very small in size, display a higher gene density than the X chromosome and the average of seven genes per Mb on autosomes. The male recombination activity in PAR1 is much higher than in the autosomes. The female recombination activity is within the autosomal range for PAR1 and probably also for PAR2. In 493 female meiosis, no recombinant has been observed in PAR2 (based on 48 families from the Kong et al study30 , data not shown). The approximate 95% confidence interval42 extends from 0.0000 to 0.0093 for the female recombination rate corresponding to an estimated recombination activity of 0–2.8 cM/Mb. The sequence of PAR2 is completely known, however in PAR1 six gaps with an estimated combined size of 370 kb could not be filled up to now.11, 43, 44
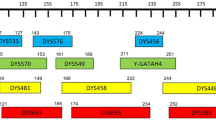
Physical locations of loci within PAR1 from different available SNP chips and genetic mapping projects are shown in Figure 2. In some studies markers sets are sparse and do not offer a good coverage of the telomeric region. In Figure 2 one can notice some larger regions that are not covered by SNPs, and these regions correspond to the sequence gaps. Estimated genetic map lengths in men vary in different studies between 12 and 55 cM in PAR1 and between 0.3 and 1.6 cM in PAR2 (Table 1). The very small value of PAR1 in the Rutgers map is because the most telomeric marker is still approximately 900 kb off the telomere. The ratio of male to female total map length in PAR1 is approximately 10, varying in single studies between 2.8 and 14.6. The deCODE map is not included because the PARs are not covered. The Marshfield map harbors few markers in PAR1 and one in PAR2 but at the time of map estimation the order of the markers was not well determined and, therefore, the map was omitted.

Physical location (build 35.1) for genetic markers in PAR1 from different mapping projects. Affymetrix and Illumina are represented by the Genome-Wide Human SNP Array 5.0 and Humhap550, respectively. Single nucleotide polymorphisms (SNPs) are shown as triangles with peak up and short tandem repeats (STRs) as triangles with peak down, squares indicate markers within the genes CSF2RA and MIC2.
Figure 3 illustrates three selected male genetic maps in PAR1 in more detail: the Duffy map45 (sperm typing), the HapMap34 map (unrelated individuals), and the map created by Henke et al6 (CEPH three-generation families). The sex-averaged estimate from HapMap was converted to a male map by using a male/female map ratio of 10:1. All maps are well in concordance for the first 750 kb whereas for the last 750 kb the HapMap estimate is much lower. This could indicate a systematic map estimation bias, a varying male/female map ratio, data errors, or random differences.

Interference
In the first genetic studies no apparent double crossover events in male meioses were observed and the question was raised whether multiple crossover events can occur at all within human PAR1.7, 9 In larger studies however, male double recombinants were found: 1 in 330 meiosis by Rappold et al,38 3 among 555 single sperms by Schmitt et al,32 and 21 among 1912 single sperms by Lien et al.33 The expected number under the assumption of no interference was 12, 15, and 177, respectively. Under Kosambi's assumption about interference, nearly 96 double recombinants would have been expected in Lien's data, significantly more than the observed number. We conclude that the absence of interference (I=0) can be rejected. Kosambi's assumption does not fit well in PAR1 and on the other hand there is no complete interference (I=1) since double recombinants have been observed. The estimated interference, I=0.96 (calculated from Lien's data) is very close to 1 indicating that the identity (1% recombination rate corresponds to 1 cM) might be the most suitable mapping function for PAR1.
Using one polymorphic marker in PAR2 and sex it has been shown that crossover events in PAR2 are possible and occur in about 2% of male meioses,2 no recombinants have been observed for PAR2 in women. In PAR2, male recombination rate is higher than in female and higher than the autosomal average but lower than in PAR1 (Table 1). This has been confirmed by other groups.30, 39
Linkage disequilibrium
In PAR1, LD and the corresponding block structures have not been analyzed in detail. Cox et al47 explored the LD in PAR1 and the remainder of the X chromosome with only seven SNPs in PAR1 and found a significant difference in LD decay. May et al48 analyzed an interval of 43 kb around the SHOX gene. Using 61 SNPs they found a rapid decline of LD, markers displaying only very low pairwise LD, and the largest block of high LD, D′>0.8, being only about 3 kb long.
In PAR2, LD has been studied with tree markers that show high allelic association between each other but not with sex-specific loci on X and Y.35 Given the male recombination rate of about 2%, this result is surprising since LD is reduced exponentially with the number of generations.
The obligate crossover
In 1982, Burgoyne1 proposed that in analogy to the autosomes a mechanism exists that ensures at least one chiasma per bivalent in PAR1, and the small size of PAR1 should prevent the formation of more than one chiasma. Later, this was referred to as ‘The theory of the single obligatory crossover in PAR1’. Since double recombinants – although very few – have been observed in different independent studies, therefore the term ‘single obligatory’ is not appropriate in general anymore. For a chromosomal region, the existence of an obligate crossover is postulated if the estimated recombination rate is 50%, meaning that there is at least one crossover. Most mapping studies are in support for this for PAR1 but not for PAR2.
It is important to keep in mind that crossing over takes place in the four chromatid bundles. If there is independent segregation, each of the four resulting chromatides has the probability 0.25 to be transmitted to an offspring. Since we can assume that in the majority of cases exactly one crossover occurs in PAR1, on average nearly 50% of the sons inherit the Y PAR1 haplotype from their fathers as a whole without recombinations.
Statistical gene localization methods
Pedigree patterns and the mode of inheritance for autosomal and X-linked Mendelian diseases can be estimated by classical segregation analysis with a large sample of families. However, the segregation pattern of loci in the PARs depends on their location relative to the pseudoautosomal boundary (PAB). Two extreme situations for an affected father are depicted in Figure 4. In situation I, the gene is close to the PAB and no recombination occurs between the gene and the sex-specific region. In II, the gene is at the telomere of PAR1 and recombines independently with a locus at the PAB. The dotted line indicates a disease mutation that could be at (1) Y-PAR1 or (2) X-PAR1. In situation I, all sons are affected if the disease mutation is on the Y-PAR1 and all daughters if the mutation is on the X-PAR1. Situation II results in pedigree patterns as for any autosomal-dominant disease. Dominant diseases caused by other PAR1 loci behave in-between these two extreme situations and loci in PAR2 behave rather as in situation I. Tests for pseudoautosomal segregation require large data sets particularly in case of the existence of phenocopies and reduced penetrance.

Pseudoautosomal segregation for a dominant disease depending on the localization of the disease gene I: near the pseudoautosomal boundary of PAR1, and II: at the telomere of PAR1. Dotted line indicates a disease allele.
Two strategies exist to determine the approximate chromosomal location for susceptibility genes, namely linkage analysis and genetic association analysis. Linkage analysis relies on the cosegregation of stretches of DNA in families broken by recombination events. It is performed by looking for cosegregation of loci with an unknown disease locus that has influenced the pattern of affected and nonaffected relatives. Association studies do not need families and aim to detect association between a marker allele and an unknown disease susceptibility allele and are often based on the comparison of unrelated cases and controls.49, 50 These statistical approaches have been implemented in several computer programs and can be used for analysis of autosomal and X-linked markers.51
Linkage analyses can be performed by using panels of microsatellites marker with different densities of 2 (2000 markers), 4 (1000 markers), or 8 cM (500 markers), usually pseudoautosomal markers are not included.52 Nowadays, genome-wide linkage and association analysis are carried out with SNPs. Table 3 shows the number of loci in PARs available on SNP chips, Affymetrix 10k and Illumina V are designed for linkage analyses.
Linkage analysis
Linkage methods are divided into so-called parametric methods for which a genetic model is assumed and nonparametric methods without assumption of a genetic model. Parametric linkage methods have been very successful for monogenic diseases and complex diseases with major genes but they require the specification of a mode of inheritance. However, for complex genetic diseases presumably caused by multiple genetic and environmental factors, the genetic model is unknown and nonparametric methods are desirable. They focus on the number of alleles shared between pairs of affected relatives. When affected relatives share more alleles identity by descent than expected by chance, this may indicate the presence of a susceptibility gene. The so-called multipoint linkage analysis uses all available markers on a chromosome and needs to assume a genetic map. Genetic distances are converted into recombination probabilities to estimate haplotypes, to find whether a disease locus cosegregates in a family.
Ott53 proposed to perform parametric linkage analysis for loci within PAR1 as in autosomes using sex-specific maps, and for sex-specific and pseudoautosomal loci together different penetrances have to be formulated according to sex and region. It can be performed with every linkage program able to handle sex-specific genetic maps. Strauch et al54, 55 suggested a recording scheme in an autosomal setting that uses codominant loci and a diallelic trait. It can be used for X-specific loci only or combined X-specific and PAR1 loci, but it requires software that offers modeling for imprinting. Both methods consider loci in PAR1, but they could also be applied to loci in PAR2.
Dupuis and Van Eerdewegh56 developed a nonparametric linkage method for loci on the PARs. It uses the concept of the expected number of alleles shared identity by decent (IBD) between affected sib pairs dependent on the sex of the sib pair. For autosomal loci, not linked to a disease, a sib pair will share 0, 1, or 2 alleles IBD with a probability of 1/4, 1/2, and 1/4, respectively. For loci on the PARs, a sib pair will always share 0 or 1 allele IBD from the maternal side with a probability of 1/2. However, the probability of sharing 0 or 1 alleles IBD from the paternal side depends on the male recombination fraction between the locus and the X-specific region and on the sex of the sib pair. For example, if a locus is located close to or right at the PAB, a pair with same sex shares at least one allele with probability 1. The probability that a mixed pair shares two alleles IBD is 0. This method does not use sex-specific maps and it is not implemented in any of the existing programs for linkage analysis. It has been shown that not taking into account the expected increase/decrease in sharing in the PAR may result in loss of power or increase in type I error rates, depending on whether there is an excess of sex-concordant pairs or excess of sex-discordant pairs.56
It is known that using sex-averaged maps in regions of sex differences leads to a reduction of power and an increase of false-positive results, even more intensively when the map density is relatively low (<1 marker per 5 cM) or the number of informative meiosis is different between sexes, as it is in the PARs.57, 58, 59 Within the PAR1 these results are particularly important since the male to female map ratio may even be higher than 10.
In the majority of the genome-wide studies, the PARs are excluded from the analysis. This may be due to the lack of reliable genetic maps in PARs, or because the analytical packages cannot deal with PARs and sex-specific loci together, or do not support sex-specific maps. Among the most widely used linkage programs, MERLIN60 and ALLEGRO61 are able to use sex-specific genetic maps, and very recently this feature has been also implemented in the GENEHUNTER-MODSCORE62 program. For more information we refer to the list of genetic analysis programs.51
Genetic association analysis
In genetic association studies, statistical tests are carried out to detect a nonrandom association between genetic polymorphisms and the trait of interest. Besides confounding and bias from several sources (eg population structure and cryptic relatedness and data errors), statistically significant genetic association can only be found if the tested polymorphism has a variant that is causally related to the disease process or if it is in strong LD with a causal variant. The latter implicates a very close location in the genome. Since LD on the population level only operates over short distances, a dense set of markers within the region of interest has to be used. As a simple rule at least one tagSNP every 5 kb was recommended to capture most of the common variation in a European sample.34 This would result in 540 common (MAF>5%) SNPs in PAR1 and 60 SNPs in PAR2. However, in PAR1 this might not be enough since the LD in PAR1 might be lower than in autosomal regions of the same physical size. Thus, 540 SNPs would constitute a low estimate. Very recently, a genome-wide significant association of one SNP in PAR1 near the CSF2RA (colony-stimulating factor receptor 2-α) with schizophrenia was found in a 500 kb SNP screen.63 However, the analysis of the LD structure in the target region within PAR1 was limited by the poor coverage of the Affymetrix 500k SNP chip. Haplotype analysis with more densely spaced SNPs in human PARs will further clarify this question.
It has not been analyzed so far whether it would be an advantage to use special analytical methods for pseudoautosomal markers in association analysis. Evidently, false-positive results could arise if a causal variant is strongly associated with male sex, and the sex distribution is different among cases and controls.
Conclusions
The human PARs have drawn considerable interest from researchers in cytogenetics, cytology, evolutionary biology, and developmental genetics. However, they have been widely ignored in linkage and genetic association studies. Maybe this is the reason why among the 29 genes known to date only 1, the SHOX gene, has been clearly correlated with disease. Recent advances in large-scale LD analysis and SNP chip technology have forwarded genome-wide association studies for complex diseases. However, there is still a paucity of SNPs in the PARs. In addition, the PARs have been largely neglected in genetic mapping studies directed toward map construction. The differences between male and female genetic maps, which are highly variable in genomic regions, chromosomal position, and population under study, are a challenge to genetic map construction in diploid organisms in which sex is determined by a pair of different sex chromosomes. For genome-wide studies, it is absolutely necessary to include a sufficient number of pseudoautosomal markers since such expensive studies should not fail to screen pseudoautosomal linkage or association. Tailored statistical methods and their integration in software tools will facilitate this purpose.
References
Burgoyne PS : Genetic homology and crossing over in the X and Y chromosomes of Mammals. Hum Genet 1982; 61: 85–90.
Freije D, Helms C, Watson MS, Donis-Keller H : Identification of a second pseudoautosomal region near the Xq and Yq telomeres. Science 1992; 258: 1784–1787.
Gabriel-Robez O, Rumpler Y, Ratomponirina C et al: Deletion of the pseudoautosomal region and lack of sex–chromosome pairing at pachytene in two infertile men carrying an X;Y translocation. Cytogenet Cell Genet 1990; 54: 38–42.
Mohandas TK, Speed RM, Passage MB, Yen PH, Chandley AC, Shapiro LJ : Role of the pseudoautosomal region in sex–chromosome pairing during male meiosis: meiotic studies in a man with a deletion of distal Xp. Am J Hum Genet 1992; 51: 526–533.
Devaux P, Kilian A, Kleinhofs A : Comparative mapping of the barley genome with male and female recombination-derived, doubled haploid populations. Mol Gen Genet 1995; 249: 600–608.
Henke A, Fischer C, Rappold GA : Genetic map of the human pseudoautosomal region reveals a high rate of recombination in female meiosis at the Xp telomere. Genomics 1993; 18: 478–485.
Page DC, Bieker K, Brown LG et al: Linkage, physical mapping, and DNA sequence analysis of pseudoautosomal loci on the human X and Y chromosomes. Genomics 1987; 1: 243–256.
Rouyer F, Simmler MC, Johnsson C, Vergnaud G, Cooke HJ, Weissenbach J : A gradient of sex linkage in the pseudoautosomal region of the human sex chromosomes. Nature 1986; 319: 291–296.
Rouyer F, Simmler MC, Vergnaud G et al: The pseudoautosomal region of the human sex chromosomes. Cold Spring Harb Symp Quant Biol 1986; 51 (Part 1): 221–228.
Ross MT, Grafham DV, Coffey AJ et al: The DNA sequence of the human X chromosome. Nature 2005; 434: 325–337.
Blaschke RJ, Rappold G : The pseudoautosomal regions, SHOX and disease. Curr Opin Genet Dev 2006; 16: 233–239.
Charlesworth D, Charlesworth B, Marais G : Steps in the evolution of heteromorphic sex chromosomes. Heredity 2005; 95: 118–128.
Lahn BT, Pearson NM, Jegalian K : The human Y chromosome, in the light of evolution. Nat Rev Genet 2001; 2: 207–216.
Graves JA : Sex chromosome specialization and degeneration in mammals. Cell 2006; 124: 901–914.
Muller HJ : A gene for the fourth chromosome of Drosophila. J Exp Zool 1914; 17: 325–336.
Aitken RJ, Marshall Graves JA : The future of sex. Nature 2002; 415: 963.
Haldane JBS : Some natural populations of Lythrum salicaria. J Genet 1936; 32: 393–397.
Moses MJ, Counce SJ, Paulson DF : Synaptonemal complex complement of man in spreads of spermatocytes, with details of the sex chromosome pair. Science 1975; 187: 363–365.
Pearson PL, Bobrow M : Definitive evidence for the short arm of the Y chromosome associating with the X chromosome during miosis in the human male. Nature 1970; 226: 959–961.
Morgan TH : Random segregation versus coupling in Mendelian inheritance. Science 1911; 34: 384.
Müller HJ : The mechanism of crossing over. Am Nat 1916; 50: 193–207.
Kosambi DD : The estimation of map distances from recombination values. Ann Eugen 1944; 12: 172–175.
Sturt E : A mapping function for human chromosomes. Ann Hum Genet 1976; 40: 147–163.
Sturt E, Smith CA : The relationship between chromatid interference and the mapping function. Cytogenet Cell Genet 1976; 17: 212–220.
McPeek MS, Speed TP : Modeling interference in genetic recombination. Genetics 1995; 139: 1031–1044.
Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A : Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 2001; 69: 831–843.
Liu BH : Statistical Genomics: Linkage, Mapping, and QTL Analysis. Boca Raton, New York: CRC Press LLC, 1997.
Broman KW, Murray JC, Sheffield VC, White RL, Weber JL : Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 1998; 63: 861–869.
Kong A, Gudbjartsson DF, Sainz J et al: A high-resolution recombination map of the human genome. Nat Genet 2002; 31: 241–247.
Kong X, Murphy K, Raj T, He C, White PS, Matise TC : A combined linkage – physical map of the human genome. Am J Hum Genet 2004; 75: 1143–1148.
Duffy DL : An integrated genetic map for linkage analysis. Behav Genet 2006; 36: 4–6.
Schmitt K, Lazzeroni LC, Foote S et al: Multipoint linkage map of the human pseudoautosomal region, based on single-sperm typing: do double crossovers occur during male meiosis? Am J Hum Genet 1994; 55: 423–430.
Lien S, Szyda J, Schechinger B, Rappold G, Arnheim N : Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am J Hum Genet 2000; 66: 557–566.
Consortium IH : A haplotype map of the human genome. Nature 2005; 437: 1299–1320.
Li L, Hamer DH : Recombination and allelic association in the Xq/Yq homology region. Hum Mol Genet 1995; 4: 2013–2016.
Arnheim N, Li H, Cui X : Genetic mapping by single sperm typing. Anim Genet 1991; 22: 105–115.
Lazzeroni LC, Arnheim N, Schmitt K, Lange K : Multipoint mapping calculations for sperm-typing data. Am J Hum Genet 1994; 55: 431–436.
Rappold GA, Klink A, Weiss B, Fischer C : Double crossover in the human Xp/Yp pseudoautosomal region and its bearing on interference. Hum Mol Genet 1994; 3: 1337–1340.
Li N, Stephens M : Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 2003; 165: 2213–2233.
McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P : The fine-scale structure of recombination rate variation in the human genome. Science 2004; 304: 581–584.
www.ensembl.org: release 45.
Agresti A, Coull BA : Approximate is better than ‘exact’ for interval estimation of binomial proportions. Amer Statist 1998; 52: 119–126.
Ross MT, Bentley DR, Tyler-Smith C : The sequences of the human sex chromosomes. Curr Opin Genet Dev 2006; 16: 213–218.
genome.ucsc.edu: UCSC Genome Browser on Human Mar, 2006, Assembly 2006.
Duffy DL : http://www.qimr.edu.au/davidD/pseudoautosomal_map.daupdate200606182006.
Consortium H : http://www.hapmap.org/downloads/recombination/latest/rates/20060920, 2006.
Cox ME, Campbell JK, Langefeld CD : An exploration of sex-specific linkage disequilibrium on chromosome X in Caucasians from the COGA study. BMC Genet 2005; 6 (Suppl 1): S81.
May CA, Shone AC, Kalaydjieva L, Sajantila A, Jeffreys AJ : Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat Genet 2002; 31: 272–275.
Thomas D : Statistical Methods in Genetic Epidemiology. New York: Oxford University Press, 2004.
Ziegler A, König IR : A Statistical Approach to Genetic Epidemiology. Weinheim: Wiley-VCH, 2006.
www.decode.com, 2007.
Ott J : Y-linkage and pseudoautosomal linkage. Am J Hum Genet 1986; 38: 891–897.
Strauch K, Baur MP, Wienker TF : A recoding scheme for X-linked and pseudoautosomal loci to be used with computer programs for autosomal LOD-score analysis. Hum Hered 2004; 58: 55–58.
Strauch K, Fimmers R, Kurz T, Deichmann KA, Wienker TF, Baur MP : Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am J Hum Genet 2000; 66: 1945–1957.
Dupuis J, Van Eerdewegh P : Multipoint linkage analysis of the pseudoautosomal regions, using affected sibling pairs. Am J Hum Genet 2000; 67: 462–475.
Halpern J, Whittemore AS : Multipoint linkage analysis. A cautionary note. Hum Hered 1999; 49: 194–196.
Daw EW, Thompson EA, Wijsman EM : Bias in multipoint linkage analysis arising from map misspecification. Genet Epidemiol 2000; 19: 366–380.
Fingerlin TE, Abecasis GR, Boehnke M : Using sex-averaged genetic maps in multipoint linkage analysis when identity-by-descent status is incompletely known. Genet Epidemiol 2006; 30: 384–396.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin – rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A : Allegro version 2. Nat Genet 2005; 37: 1015–1016.
Dietter J, Mattheisen M, Furst R, Ruschendorf F, Wienker TF, Strauch K : Linkage analysis using sex-specific recombination fractions with GENEHUNTER-MODSCORE. Bioinformatics 2007; 23: 64–70.
Lencz T, Morgan TV, Athanasiou M et al: Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry 2007; 12: 572–580.
Acknowledgements
AF was supported by a grant of the Nationales Genomforschungsnetz NGFN-01GR0465. We thank T Matise for providing us with genotype data for the pseudoautosomal regions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flaquer, A., Rappold, G., Wienker, T. et al. The human pseudoautosomal regions: a review for genetic epidemiologists. Eur J Hum Genet 16, 771–779 (2008). https://doi.org/10.1038/ejhg.2008.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.63
Keywords
This article is cited by
-
Genome-wide single nucleotide polymorphism array analysis unveils the origin of heterozygous androgenetic complete moles
Scientific Reports (2019)
-
Preferential Y-Y pairing and synapsis and abnormal meiotic recombination in a 47,XYY man with non obstructive azoospermia
Molecular Cytogenetics (2016)
-
Back to the roots: segregation of univalent sex chromosomes in meiosis
Chromosoma (2016)
-
Initiation of recombination suppression and PAR formation during the early stages of neo-sex chromosome differentiation in the Okinawa spiny rat, Tokudaia muenninki
BMC Evolutionary Biology (2015)
-
Genomic identification and characterization of the pseudoautosomal region in highly differentiated avian sex chromosomes
Nature Communications (2014)
