
Abstract
The aim of this study was to identify and characterize the underlying molecular mechanisms in autosomal-dominant retinitis pigmentosa (adRP) with incomplete penetrance in two Swedish families. An extended genealogical study and haplotype analysis indicated a common origin. Mutation identification was carried out by multiplex ligation-dependent probe amplification (MLPA) and sequencing. Clinical examinations of adRP families including electroretinography revealed obligate gene carriers without abnormalities, which indicated incomplete penetrance. Linkage analysis resulted in mapping of the disease locus to 19q13.42 (RP11). Sequence analyses did not reveal any mutations segregating with the disease in eight genes including PRPF31. Subsequent MLPA detected a large genomic deletion of 11 exons in the PRPF31 gene and, additionally, three genes upstream of the PRPF31. Breakpoints occurred in intron 11 of PRPF31 and in LOC441864, ‘similar to osteoclast-associated receptor isoform 5.’ An almost 59 kb deletion segregated with the disease in all affected individuals and was present in several asymptomatic family members but not in 20 simplex RP cases or 94 healthy controls tested by allele-specific PCR. A large genomic deletion resulting in almost entire loss of PRPF31 and three additional genes identified as the cause of adRP in two Swedish families provide an additional evidence that mechanism of the disease evolvement is haploinsufficiency. Identification of the deletion breakpoints allowed development of a simple tool for molecular testing of this genetic subtype of adRP.
Similar content being viewed by others
Introduction
Retinitis pigmentosa (RP) is a group of inherited retinal disorders with a considerable genetic variation. Typical signs of the disease are night blindness and progressive loss of the peripheral visual field, characteristic pigment deposition in the retina, attenuation of the retinal blood vessels, and optic disc pallor.
A form of adRP in patients with night blindness in the first and second decade, progressive visual field loss in later life, along with asymptomatic individuals though having an affected parent and an affected child was mapped to 19q13.41, 2 (RP11, MIM 600138). Later, a putative human ortholog of yeast pre-mRNA splicing factor, PRPF31, was reported as a disease-causing gene.3 To date, 42 PRPF31 mutations are listed in the Human Genome Mutation Database and among publically available only 9 are missense, whereas the remaining are deletions (16), insertions (2), indels (2), and splicing mutations (11) (http://www.hgmd.cf.ac.uk/ac/all.php).The majority of mutations would result in truncated proteins because of exon skipping and premature stop codons;3, 4, 5, 6, 7 therefore, haploinsufficiency was suggested as a mechanism of RP11 evolvement.3 The size of the PRPF31 deletions varies significantly from a single nucleotide to a deletion larger than 45 kb.3, 4, 5, 6, 7, 8, 9 Large genomic deletions including PRPF31 account for 2.5% of adRP cases.6
In this study of two families with adRP linked to 19q13.42, we identified a novel genomic deletion including almost the entire PRPF31 gene. One family showed complete disease penetrance, whereas in another family asymptomatic disease gene carriers were present. On the basis of defined breakpoint sequences, we developed allele-specific PCR for molecular testing of adRP patients.
Materials and methods
Patients and ophthalmologic examinations
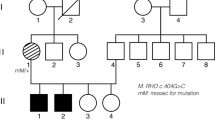
On the basis of genealogical studies back to the beginning of the 18th century, we created two large pedigrees 008 (Figure 1) and 078 (Figure 2), both traced to a small village (Klabböle). Blood samples and informed consent were obtained from 44 individuals from the families (7 affected among totally 11 in the family 008 and 12 affected among totally 33 in the family 078) and 20 simplex cases. The study was approved by Ethics Committee at University Hospital of Umeå followed the tenets of the Declaration of Helsinki Principles and by the Committee for Protection of Human Subjects, The University of Texas (Health Science Center, Houston, TX, USA). Standard ophthalmologic examination included fundus photography and visual field testing. Dark adaptation tests and full-field ERGs were performed in selected cases.

Haplotype analysis (a) and segregation of the mutation (b) in family 008. (a) Filled symbols indicate affected individuals, whereas empty symbols indicate unaffected. Only disease haplotypes shared by affected individuals in both families are boxed. Microsatellite markers and their order are described in Materials and Methods. (b) Allele-specific PCR where a band of 385 bp indicates the mutant allele and a band of 101 bp indicates presence of the internal PCR control, as a result of wild-type and mutant alleles amplification.

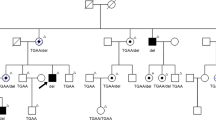
Haplotype analysis (a) and segregation of the mutation (b) in the family 78. (a) Filled symbols indicate affected individuals, whereas empty symbols indicate unaffected. Symbols with · represent an asymptomatic gene carrier. Only disease haplotypes shared by affected individuals in both families are boxed. Microsatellite markers and their order are described in Materials and Methods. (b) Allele-specific PCR where a band of 385 bp indicates the mutant allele and a band of 101 bp indicates presence of the internal PCR control, as a result of wild-type and mutant alleles amplification.
Molecular genetic analysis
Genotyping, linkage, and sequencing
DNA was extracted from peripheral blood and used for linkage analysis with the ABI PRISM Linkage Mapping Set (version 2.5; Applied Biosystems, Foster City, CA, USA) as described elsewhere.10 Fine mapping and haplotype analysis were performed with 10 microsatellite markers (D19S888, D19S921, D19S572, D19S924, D19S927, D19S926, D19S418, D19S605, D19S891, and D19S210) according to Köhn et al.10 Two-point linkage analyses were performed using the FASTLINK implementation of the LINKAGE program package.11 A penetrance value of 0.7 (the ratio between symptomatic individuals and all disease-gene carriers) was used to account for incomplete penetrance.
Sequence analysis of coding exons and adjacent intronic sequences of candidate genes (RDH13, PPP1R12C, SYT5, PRKCG, CACNG6, 7, 8, and PRPF31) was performed as described by Köhn et al10 (primers sequences are available upon request).
MLPA and breakpoint analysis
Multiplex ligation-dependent probe amplification (MLPA) was performed according to Sullivan et al6 on two affected and two unaffected individuals from each family with a set of probes designed by this group in combination with the Retinitis Pigmentosa Kit (MRC Holland; http://www.mrc-holland.com/pages/indexpag.html). In additional, we used VSTM1 probe with following sequences: forward, 5′-GGGTTCCCTAAGGGTTGGACCTTCACGGACCTGAAGCCTAAGGATGCTGGGAG; reverse, 5′-GTACTTTTGTGCCTACAAGACAACAGCCTCCCATGAGTGGTCTAGATTGGATCTTGCTGGCAC (DNA Technology, Risskov, Denmark). The collected raw data were analyzed with ABI Prism GeneMapper Software v3.0 (Applied Biosystems). Calculation of probe copy numbers was based on Stern et al.12 The ratio of 1.0 indicates the presence of two alleles (normal diploid) and 0.5 or 1.5 suggests either deletion or duplication of the target sequence, respectively.
The breakpoint region was defined using long-range PCR (Qiagen) across the deletion with the following primers: forward VSTM F1, 5′-GATAGAGGAGGTTTTGCTCTGAC and reverse PRPF31 13R, 5′-CGGACCCTGCAGAAGCAGAGCGTCGTAT. PCR product was cloned into pGEM-T Easy (Promega) vector and positive clones were sequenced.
Allele-specific PCR
PCR on genomic DNA was performed as described elsewhere using specific primers for both mutant and wild-type alleles (BP-F: 5′-TGAAAGAGAGAAGGGGCTCA; BP-R: 5′-GTGGCCTCGTTTACCTGTGT; and cDNA PRPF31 12F: 5′ATCGAGGAGGACGCCT).
Results and discussion
Clinical findings
The initial symptom in all patients was difficulty in seeing at night from school or pre-school age. The age at diagnosis was remarkably high, 40–50 years, in the majority of the cases. The two youngest patients were diagnosed at the age of 15 and 17 years, respectively. The clinical phenotype could be referred to as a typical or classical RP with bone spicule changes and a considerable variation in severity. Some patients had a quite preserved visual field with moderate losses in the mid-periphery at the age of 50 years, whereas other cases only had a central field of 5–10° at the age of 37–45 years. All patients with clinical symptoms presented non-recordable rod responses when examined electrophysiologically. Patients with only minor defects in the mid-periphery of the visual field showed severely reduced rod-cone responses, but normal amplitudes of the cone responses. The implicit times were prolonged. In patients with more severe visual field defects, mixed rod-cone or cone responses were not recordable. One of the non-symptomatic carriers (Case VII:15) was examined with ERG at the age of 50 and 60 years with normal recordings.
Molecular findings
Two Swedish families (008 and 078) from the same geographical region showed an autosomal-dominant inheritance pattern. The father to V:1 (family 008) was asymptomatic; however, we did not have his DNA for any analyses (Figure 1a). In the family 078, at least two individuals were obligate carriers of the mutation, as they had both an affected parent and offspring (VII:8, VII:15) (Figure 2a).
Significant LOD scores with a maximum of 7.58 at the marker D19S926 were revealed in the region spanning nearly 1.77 Mb. The reconstructed haplotypes in both families confirmed the segregation of adRP with markers D19S924, D19S927, D19S926, D19S418, and D19S605. The adRP locus was narrowed slightly because of recombination events for D19S572 and D19S891 markers. Unaffected individual VII:18 in family 078 shared disease haplotype for markers D19S926, D19S418, and D19S605, which suggested that the causative gene is located in close proximity to D19S924 and D19S927. Coding regions and flanking intron sequences of RDH13, SYT5, PPP1R12C, PRKCG, CACNG6, 7, 8, and PRPF31 located in the refined region were analyzed by direct sequencing. As no disease-causing mutations were detected by PCR-based methods, we considered testing for large genomic deletions using MLPA examining PRPF31, RHO, RP1, RPE65, and IMPDH1 genes. A large genomic deletion including TFPT, NDUFA3, and OSCAR genes and 11 exons of the PRPF31 gene were detected (Figure 3a).

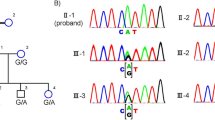
Genomic deletion including PRPF31 gene in adRP. (a) A deletion detected by MLPA in VIII:2 from the family 078. A P235 Retinitis Pigmentosa kit (MRC Holland) along with the VSTM1 probe was applied. The graph indicates the presence of one gene copy with probes for exons 1–11 in the PRPF31 gene (ratio is ∼0.5). Two gene copies are present with the probes for IMPDH1, RHO, RP1, and for exons 12–14 in the PRPF31 (ratio is ∼1.0). (b) Schematic representation of the genomic region 19q13.42 in proximity to the PRPF31. PCR primers VSTM1 1F and PRPF31 13R were applied to amplify across the deleted region (the estimated size of a wild-type allele is 65 530 bp and cannot be amplified by PCR). PCR fragment of ∼7 kb (mutant allele) was obtained by a long-range PCR, subcloned into pGEM-T Easy vector and sequenced. (c) Localization of allele-specific primers. Primers sequences and allele-specific PCR are described in Materials and Methods and Results and Discussion. PCR with allele-specific primers BP-F and BP-R primers used for segregation analysis (Figures 1b and 2b) resulted in 385 bp fragment representing a mutant allele. (d) A partial sequence across the deletion. Nucleotide sequence belonging to the predicted gene ‘similar to osteoclast-associated receptor isoform 5’ is shown in red and sequence of the breakpoint in intron 11 in the PRPF31 gene is shown in green. CAA in black can be part of either LOC441864 or PRPF31.
On the basis of MLPA results indicating a normal ratio for the VSTM1 probe and probes for exons 12–14 of the PRPF31, we applied a long-range PCR with VSTM1 and exon 13 PRPF31 primers (Figure 3b) and obtained an ∼7-kb PCR fragment. Sequencing of this product revealed deletion of 58 733 nucleotides with breakpoints in intron 11 of the PRPF31 gene and in the LOC441864 (ref∣NT_011109.15∣Hs19_11266), similar to osteoclast-associated receptor isoform 5 (Figure 3b–d).
Allele-specific PCR with primers set as shown in Figure 3c revealed the presence of the mutation in affected individuals, obligatory carriers, and also several asymptomatic members in the 078 family (Figures 1b and 2b). None of 20 simplex RP cases or 94 healthy controls (188 control chromosomes) from the matched population showed the mutant allele (data not shown).
PRPF31 codes for a protein needed for splicing in all cell types, although its pathologic effect is seen only in rod photoreceptors, causing adRP with incomplete penetrance. PRPF31 is a 61-kDa protein, part of the U4/U6-U5 tri-snRNP complex.13 The proposed mechanism of adRP (RP11) evolvement is haploinsufficiency rather than a dominant-negative effect.3 In this study, we identified a large genomic deletion with almost entire loss of PRPF31 gene, which provides additional evidence for haploinsufficiency as the mechanism in adRP pathogenesis.
Molecular methods used for mutation detection are mainly based on PCR and, therefore, large genomic rearrangements are easily missed. Genomic deletions at 19q13.42 ranging from 5 to more than 45 kb were reported earlier;6, 9 however, this is the first report on the identification of breakpoints in a large deletion.
The deletion encompasses almost 59 kb of genomic sequence, includes three additional genes and breakpoints occur in intron 11 of PRPF31 and within the predicted gene, LOC441864, annotated as ‘similar to osteoclast-associated receptor isoform 5.’ A number of Alu-repeats in PRPF31 introns are prone to internal unequal recombination, resulting in a deletion; however, the exact mechanism is not known.
Owing to the size of the deletion, we could expect a severe phenotype in our families as reported earlier.9 However, among our patients, there were individuals with quite preserved visual fields and recordable ERGs in their 50s. The phenotype presented with classical symptoms of RP was similar in both families. No additional symptoms associated with the genetic defects in NDUFA3, TFPT, or OSCAR were observed. This indicates that one copy of these genes is sufficient for cell function. It can be noted that one family showed complete penetrance, whereas in a larger family nine individuals were asymptomatic gene carriers, two of whom have one affected parent and offspring (Figure 2a, VII:8 and VII:15). Incomplete penetrance is believed to be caused by allelic imbalance when the wild-type allele is overexpressed compensating for the non-functional allele in asymptomatic gene carriers.14, 15 However, in our study (family 078), this hypothesis would not work because four gene carriers had the same wild-type allele as their affected siblings (Figure 2a).
In conclusion, identification of such large deletion involving the PRPF31 gene reveals additional evidence that haploinsufficiency is a molecular mechanism of evolvement of adRP with incomplete penetrance. Identification of deletion breakpoint provides an important tool for molecular testing and genetic counseling of these patients.
References
Moore AT, Fitzke F, Jay M et al: Autosomal dominant retinitis pigmentosa with apparent incomplete penetrance: a clinical, electrophysiological, psychophysical, and molecular genetic study. Br J Ophthalmol 1993; 77: 473–479.
Al-Maghtheh M, Inglehearn CF, Keen TJ et al: Identification of a sixth locus for autosomal dominant retinitis pigmentosa on chromosome 19. Hum Mol Genet 1994; 3: 351–354.
Vithana EN, Abu-Safieh L, Allen MJ et al: A human homolog of yeast pre-mRNA splicing gene, PRPF31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell 2001; 8: 375–381.
Martinez-Gimeno M, Gamundi MJ, Hernan I et al: Mutations in the pre-mRNA splicing-factor genes PRPF3, PRPF8, and PRPF31 in Spanish families with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2003; 44: 2171–2177.
Sato H, Wada Y, Itabashi T, Nakamura M, Kawamura M, Tamai M : Mutations in the pre-mRNA splicing gene, PRPF31, in Japanese families with autosomal dominant retinitis pigmentosa. Am J Ophthalmol 2005; 140: 537–540.
Sullivan LS, Bowne SJ, Seaman CR et al: Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2006; 47: 4579–4588.
Ghazawy S, Springell K, Gauba V, McKibbin MA, Inglehearn CF : Dominant retinitis pigmentosa phenotype associated with a new mutation in the splicing factor PRPF31. Br J Ophthalmol 2007; 91: 1411–1413.
Wang L, Ribaudo M, Zhao K et al: Novel deletion in the pre-mRNA splicing gene PRPF31 causes autosomal dominant retinitis pigmentosa in a large Chinese family. Am J Med Genet 2003; 121: 235–239.
Abu-Safieh L, Vithana EN, Mantel I et al: A large deletion in the adRP gene PRPF31: evidence that haploinsufficiency is the cause of disease. Mol Vis 2006; 12: 384–388.
Köhn L, Kadzhaev K, Burstedt MSI et al: Mutation in PYK2-binding domain of the PITPNM3 causes autosomal dominant cone dystrophy (CORD5). Eur J Hum Genet 2007; 15: 664–671.
Weeks DE, Ott J, Lathrop GM : SLINK: a general simulation program for linkage analysis. Am J Hum Genet 1990; 47: A204 (supplement).
Stern RF, Roberts RG, Mann K, Yau SC, Berg J, Ogilvie CM : Multiplex ligation-dependent probe amplification using a completely synthetic probe set. BioTechniques 2004; 37: 399–405.
Makarova OV, Makarov EM, Liu S, Vornlocher HP, Lührmann R : Protein 61K, encoded by a gene (PRPF31) linked to autosomal dominant retinitis pigmentosa, is required for U4/U6*U5 tri-snRNP formation and pre-mRNA splicing. EMBO J 2002; 21: 1148–1157.
Rivolta C, McGee TL, Rio Frio T, Jensen RV, Berson EL, Dryja TP : Variation in retinitis pigmentosa-11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with dominant RP11 mutations. Hum Mutat 2006; 27: 644–653.
Vithana EN, Abu-Safieh L, Pelosini L et al: Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci 2003; 44: 4204–4209.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Köhn, L., Bowne, S., S Sullivan, L. et al. Breakpoint characterization of a novel ∼59 kb genomic deletion on 19q13.42 in autosomal-dominant retinitis pigmentosa with incomplete penetrance. Eur J Hum Genet 17, 651–655 (2009). https://doi.org/10.1038/ejhg.2008.223
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.223
