
THE PAPER IN BRIEF
• Ultrashort, intense X-ray pulses generated at facilities known as X-ray free-electron lasers (XFELs) have been used to probe light-induced structural changes in proteins.
• Light-responsive proteins typically absorb one optical or ultraviolet photon in natural settings, but could absorb more from the intense ‘pump’ lasers used to induce structural changes in these studies.
• Such unnatural absorption of multiple pump photons might force proteins to behave in ways that are not biologically relevant.
• Questions have therefore been raised about how these studies should be interpreted.
• Barends et al.1 now show that the structure of a model protein changes in different ways depending on whether single or multiple photons are absorbed.
RICHARD NEUTZE: Imperfect experiments can be informative
Structural changes that occur in proteins during biochemical reactions can be measured using a technique called time-resolved X-ray diffraction (TR-XRD). In this method, reactions are initiated in protein crystals, and X-ray pulses are used to record X-ray diffraction data at selected times after initiation. TR-XRD has produced structural insights into the pathways of diverse biological processes2, including photosynthesis, sensory signalling, ion transport and photodissociation — the light-induced breakage of bonds between proteins and their ligand molecules.
Read the paper: Influence of pump laser fluence on ultrafast myoglobin structural dynamics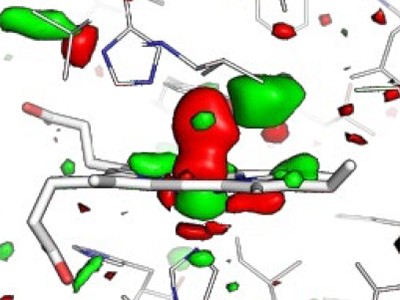
For light-sensitive proteins, a pump laser pulse is used to initiate the reaction of interest. All molecules probed in a crystal contribute to the measured X-ray diffraction pattern, yet typically only a subpopulation is activated by the pump laser. A quantity known as the crystallographic occupancy estimates the fraction of molecules in a crystal that are activated. Raising the pump-laser fluence — the energy delivered per unit area by the pump laser onto a crystal — can increase the crystallographic occupancy, but more than one photon can be absorbed by the protein at high laser fluences3,4.
Barends et al. studied structural changes that occur in the carbon monoxide complex of the protein myoglobin (MbCO) after pump-laser-induced photodissociation of CO from the iron atom of a haem group (Fig. 1). This process was previously studied using TR-XRD at time resolutions of 7.5 nanoseconds5 and 150 picoseconds (1 ps is 10–12 seconds)6 using relatively large protein crystals (dimensions in the range of about 0.1 to 0.3 millimetres) and X-ray pulses from a synchrotron facility, which is a less intense X-ray source than an XFEL.

Figure 1 | Structural dynamics of a model protein system. Barends et al.1 used a method called time-resolved X-ray diffraction (TR-XRD) to study crystals of the carbon monoxide complex of the protein myoglobin (MbCO; only part of the complex is shown). They used a ‘pump’ laser pulse to induce photodissociation (breakage of the bond between CO and the iron (Fe) atom of a haem group in the protein), and then used ultrashort, intense X-ray pulses to obtain X-ray structures of the protein over time. Three structural changes crucial to photodissociation are shown (right): flexing of the haem group; changes in the distance between the haem and a histidine amino-acid residue in the F α-helix of the protein; and displacement of the F-helix. The authors observed that the fluence of the pump laser (the energy delivered per unit area by the pump laser onto a crystal) alters the amplitude and timing of these motions, and also the motion of the CO away from the haem after photodissociation — suggesting that lower fluences are needed to observe structural changes that occur in natural settings.
A 2015 study by some of the same researchers as Barends et al. used extremely short, intense XFEL pulses to record TR-XRD data from tens of thousands of much smaller MbCO crystals (average size 15 micrometres × 5 µm × 3 µm). This thereby achieved a time resolution of 250 femtoseconds (1 fs is 10–15 s) and revealed ultrafast conformational changes of the protein as photodissociation occurs7. But because those experiments used a high pump-laser fluence, Barends et al. have now repeated their study using a lower fluence that ensures single-photon excitation of MbCO.
The authors used their TR-XRD data to determine difference Fourier maps, which show differences in electron density in MbCO before and after activation. Barends et al. found that lower pump-laser fluences yield lower crystallographic occupancies in maps produced 10 ps after protein activation. For this time delay, differences between structural changes induced by single-photon and multiphoton excitation are difficult to see from the difference Fourier maps, but the authors argue that some effects can be determined from careful analysis and structural modelling. A caveat is that structural modelling incurs larger errors when the crystallographic occupancy is low.
Clues to how water splits during photosynthesis
For sub-picosecond time delays, the authors’ analysis shows that the protein responds unexpectedly rapidly to the highest pump-laser fluence, and that CO swiftly populates a new location near the haem (Fig. 1). By contrast, it takes longer for the protein to respond and for this CO site to be populated after single-photon activation.
Barends and colleagues’ recording of high-quality maps from very small crystals of MbCO after single-photon excitation is impressive. Their study will motivate other researchers to recover high-quality difference Fourier maps using single-photon excitation. However, this might not be possible for many biological systems. Crucially, the structural perturbations of MbCO induced by single photons are in keeping with findings from earlier work5–7, illustrating that useful insight does emerge from imperfect experiments. Historically, the field has chosen not to let the perfect be the enemy of the good. I contend that such pragmatism should continue, so that the diversity of biological systems studied by TR-XRD continues to grow2.
R. J. DWAYNE MILLER: Protein ‘music’ must not be distorted
Biological processes are driven by chemistry. Chemistry is dynamic, with all the interconnecting atoms in molecules jiggling around, vying for right of way to take part in reaction pathways. In proteins, chemical transformations occur at an active or binding site. These processes involve the coupling of reaction forces such that some 10–100 atoms at the active site direct the motions of the 1,000–10,000 (or more) atoms of the surrounding protein. An astronomical number of possible conformational pathways (sequences of molecular structures) can occur, but only one or a few motions at the active site — known as reaction modes — direct biological function.
How can such a small number of reaction modes preferentially direct protein functions within the ocean of alternative conformational pathways? This apparent paradox is solved by considering spatially correlated motions — these arise when forces imprinted in the protein structure cause atoms to move together, rather than randomly and independently. By directly observing atomic motions during the defining moments of a chemical reaction, the initial distillation of all possible pathways down to a few reaction modes can be seen8. Such observations allow relationships between protein structure and function to be determined, but only if the reaction is initiated correctly. The whole point of such studies is that we don’t know the length and timescales of protein responses to the chemical driving force.
To help make sense of this issue, consider the fluid that forms by mixing cornflour and water. If you dip your finger slowly into this mixture, the fluid flows around it like a liquid. But if you rapidly poke the mixture, the material responds as an elastic solid. Similarly, the spatially varying barriers to motions in proteins mean that protein responses to forces depend on both the time and the length scales of the applied force. Given that protein structures are highly anisotropic (different in different directions), the response will also depend on the spatial location of the induced force.
Earliest molecular events of vision revealed
Until the work of Barends et al., femtosecond TR-XRD studies had used extremely high pump fluences to ensure that structural changes in proteins could be observed. Because the laser pulses are so short, this came at the cost of inducing multiphoton protein activation that produces initial atomic displacements different to those arising from single-photon activation, and at higher energies, with the atoms being displaced farther from their equilibrium positions4. The driving forces for the observed structural changes were therefore both spatially different and significantly larger than those of the biological pathway of interest4.
For the model MbCO system, there are three biologically crucial and spatially coupled motions (Fig. 1): the haem doming coordinate (which describes the flexing of the protein’s haem group); movement of a histidine amino-acid residue positioned close to the haem; and the displacement of α-helices resulting from the transfer of driving forces from the first two motions. Barends et al. observed that multiphoton activation resulted in much faster displacement of the histidine than did single-photon activation, and led to similarly faster motions of one of the helices. Moreover, the spatially correlated helical motions extended over different distances along the helix than did those induced by single-photon activation, and subsequently dissipated to different degrees.
The impulsive nature of the force produced by multiphoton activation on the initial histidine motion, and the temporal evolution of the positioning of the CO molecule in the protein pocket that contains the haem, are evidence of a different reaction pathway to the one produced by single-photon activation. There are significant differences in the magnitude, temporal evolution and pathway of the structural dynamics throughout the protein.
Given the importance of energetics and spatially correlated motions for understanding the structural changes that underpin protein function, we need to get this right. Biologically relevant protein motions are like music, albeit playing at frequencies we can’t hear. We need to observe the motions that correspond to certain frequencies, and discern how those motions are damped to produce net displacements. Imagine listening to music in which violin strings are struck too strongly, causing the accidental playing of different chords together. You would not hear the music as written, but something else entirely. To understand how nature works, we need to listen to the molecular music as nature wrote it. Barends et al. have done just that.
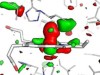 Read the paper: Influence of pump laser fluence on ultrafast myoglobin structural dynamics
Read the paper: Influence of pump laser fluence on ultrafast myoglobin structural dynamics
 Clues to how water splits during photosynthesis
Clues to how water splits during photosynthesis
 Earliest molecular events of vision revealed
Earliest molecular events of vision revealed




