THE TOPIC IN BRIEF
• Some aspects of microbiology remain mysterious because of a lack of information about the identity and role of many microbial genes and proteins.
• The ability to obtain and analyse microbial sequences at scale and across species, including those that cannot be grown under laboratory conditions, are providing insights and data to explore.
• Writing in Nature, Rodríguez del Río et al.1 report their analysis of 149,842 bacterial genomes sampled from a variety of habitats in the wild.
• The data were used to select sequences to generate a catalogue of 404,085 previously unknown gene families that could be prioritized for further study.
• The investigation of these previously unknown genes could lead to new clinical tools or offer fresh perspectives about how microorganisms evolved to survive in their natural environments.
JAKOB WIRBEL & AMI S. BHATT: Bringing structure and context to gene mysteries
The function of most microbial genes is unknown. Some of this microbial ‘dark matter’ might encode previously unknown types of enzyme or classes of antibiotic. As ever more genes of unknown function are discovered through sequencing of DNA from mixtures of multiple genomes, termed metagenomic sequencing, the difficulty of experimentally characterizing these enigmatic genes has led to a focus on computationally predicting their function2. Two publications in Nature, one by Rodríguez del Río et al.1, and one by Pavlopoulos et al.3 published last October, tackle this challenge by cleverly leveraging advances in clustering algorithms (computational tools that group genes on the basis of similarities in amino-acid sequence) and protein-structure prediction tools4 such as AlphaFold.
Read the paper: Functional and evolutionary significance of unknown genes from uncultivated taxa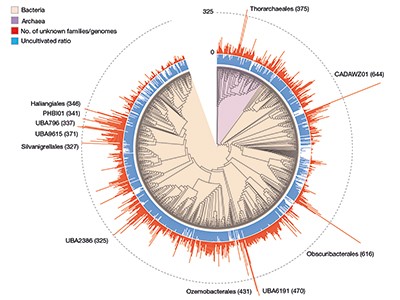
Despite distinct technical approaches, the core strategy used by Pavlopoulos et al. and Rodríguez del Río et al. was similar. Both clustered hundreds of millions of protein sequences from metagenomic data sets into previously unknown protein families. Rodríguez del Río and colleagues filtered their data to examine genes only from prokaryotes (organisms whose cells lack a nucleus), whereas Pavlopoulos et al. used data that also included sequences from eukaryotes (organisms whose cells have a nucleus) and viruses.
With these catalogues of previously unknown families at hand, both teams set out to predict the function of their newly described families, capitalizing on genomic-context analysis, which involves examining adjacent genes for clues about function, as well as harnessing breakthroughs in methods to predict protein structures. In prokaryotic genomes, genes involved in the same pathway are often present close to one other. Genomic-context analysis, which proposes ‘guilt by association’, has been used effectively to predict previously unknown antiviral defence systems used by bacteria5. The second approach, comparing predicted protein structures to find similar (homologous) proteins, is more sensitive than simply comparing amino-acid sequences alone6. Both teams predicted structures for their protein families and compared them with databases of known structures, thereby generating informed predictions about the function of some of these enigmatic proteins.
The sheer scale and computational investment involved in these efforts, which yielded hundreds of thousands of newly discovered protein families (Fig. 1), is impressive. Yet, the number of previously unknown genes that have a functional prediction still remains relatively small. In both publications, only around 15% of the previously unknown protein families could be annotated on the basis of structural similarity; genomic-context analysis enabled functions to be proposed for 7.4% of families in Pavlopoulos et al. and 13% in Rodríguez del Río and co-workers. In addition, some assigned functional categories (such as ‘ribosome’) lack detailed specificity and this might obscure the precise role of these genes. Ultimately, the reliability of these predictions will have to be determined experimentally. Indeed, Rodríguez del Río et al. took the first step towards this objective by experimentally verifying the annotation for two of their predicted families.

Figure 1 | Previously unknown microbial gene families. The large-scale analysis of DNA sequences captured from microbial samples as reported by Rodríguez del Río et al.1 and by Pavlopoulos et al.3 has revealed hundreds of thousands of previously unknown gene families. These data — which were gathered from microbes in the wild and across different habitats, and include species that have not been cultivated in the laboratory — provide a starting point for gaining insights into unexplored aspects of the biology of bacterial and archaeal microorganisms. Figure adapted from Fig. 3a of ref. 1.
By delving deeper into the microbial dark matter, these two studies unlock a wealth of previously hidden knowledge, paving the way for future discoveries in diverse fields from medicine to biotechnology. Follow-up experiments might include the study of protein families with completely new protein folds, possibly revealing unexplored biological functions. Similarly, synapomorphic genes — corresponding to protein families that are specific to a group of organisms sharing a common ancestor but absent in others — might hold clues to key evolutionary processes. With further refinement and validation, these computational approaches offer a powerful tool for unlocking the functional secrets of the unseen microbial world.
ALEXANDER J. PROBST: Microbial sequences reveal ecology and evolution
Genes are the ultimate source of all biological information on Earth, from human eye colour to the cell shape of microorganisms. The proteins they encode can be grouped using bioinformatics into families, usually with shared functionality. The ensemble of all known proteins in databases is continuously expanding as genomes are sequenced and the functions of the encoded proteins are predicted. The greatest fraction of biological functional diversity on our planet is attributed to microbial proteins. With the advent of sequencing of mixed microbial genomes from the environment (an approach that explores multiple genomes and is called metagenomics7), the increase in the rate at which data are being added to genome and protein databases is striking. However, the functional capacity of most protein families is unknown and part of the microbial dark matter.
Rodríguez del Río and colleagues’ work, as well as the study by Pavlopoulos et al., analysed large-scale metagenomic data and explored the potential function and distribution of unknown protein families, which might have evolutionary and ecological importance. Rodríguez del Río analysed nearly 150,000 microbial genomes (Fig. 1), and Pavlopoulos and colleagues investigated nearly 27,000 metagenomic data sets retrieved from diverse ecosystems with various bioinformatics approaches — going well beyond the scale of public-database entries used in previous such studies8. Surprisingly, a method called rarefaction analysis used by Pavlopoulos and colleagues revealed no slowing down in the detection of previously unknown protein families as new metagenomes were added to their analysis. Instead, the detection of protein families increased exponentially, warranting an array of follow-on studies.
The distribution of protein families across Earth’s categories of ecosystem (biomes) presented by Pavlopoulos and colleagues corroborates the findings of previous investigations regarding the distribution of microbial genes8. Some biological entities, however, were particularly rich sources of newly discovered protein families, including viruses, as Pavlopoulos et al. report, and microbes called Asgardarchaeota, as presented by Rodríguez del Río and colleagues. The latter are a group of microorganisms called archaea that are closely related to the first ancestor of eukaryotes. As such, studying their proteins might reveal new insights into the evolution of the eukaryotic cell9.
Crowdsourcing Earth’s microbes
One major challenge in exploring the wealth of previously unknown protein families encoded in genomes of natural samples is the identification of eukaryotic genes in metagenomes. Although certain algorithms exist for the recovery of eukaryotic genomes from metagenomes, accurately predicting eukaryotic genes in mixed DNA sequences — equivalent to Pavlopoulos and colleagues’ method of identifying microbial genes — is still not possible bioinformatically. Once this shortcoming is overcome with the development of new algorithms, scientists will substantially expand the protein ‘sequence space’ and will identify protein families of unknown function that drive the ecology and evolution of eukaryotes.
The greatest advance in painstakingly organizing the protein families of nearly 27,000 metagenomes and across the tree of life lies in the identification of ecosystem-specific protein clusters that differ in terms of their presence or absence, or relative abundance between varying conditions of a given ecosystem — for example, between the contexts of health or disease. Applying this strategy to examine microbial data for healthy people and those with colorectal cancer, Rodríguez del Río and colleagues found that specific unknown protein families were enriched in the gut bacteria of people with cancer. These protein families were associated with microbial motility, adhesion and invasion potentially of human tissue, as revealed through genomic-context analysis. Harnessing this approach in other fields of research should be extremely helpful for deciphering the different functions of sample sets, in the hope of identifying new targets for biochemical analyses to shed light on a tiny fraction of the microbial dark matter.
Identifying differences in microbial communities (microbiomes) that might explain, for example, the disease state of a person, rely heavily on comparing which species are present and how abundant they are (the taxonomic composition), and examining genes that are associated with certain functions. Finding specific but differentially abundant protein families of unknown function, as demonstrated by Rodríguez del Río and co-workers, has the potential not only to replace current marker-gene-based approaches for differentiating microbiomes but also to advance microbiome research to a new and causality-driven level.
 Read the paper: Functional and evolutionary significance of unknown genes from uncultivated taxa
Read the paper: Functional and evolutionary significance of unknown genes from uncultivated taxa
 Tracking humans and microbes
Tracking humans and microbes
 Crowdsourcing Earth’s microbes
Crowdsourcing Earth’s microbes