
ARYA MASSARAT & MELISSA GYMREK: Describing genetic diversity with graphs
Reference genomes are crucial coordinate systems for genomic analyses. However, the references that scientists currently work from when studying humans (the draft human genome1 and its complete, gap-free successor2, dubbed T2T-CHM13) are both based mostly on single individual genomes. A linear genome sequence of this type cannot adequately represent genetic diversity within our species. Instead, such diversity is more accurately described using a graph-based system of branching and merging paths. In a paper in Nature, Liao et al.3 describe the first human reference pangenome — a collection of genome sequences compiled into a single data structure.
Read the paper: A draft human pangenome reference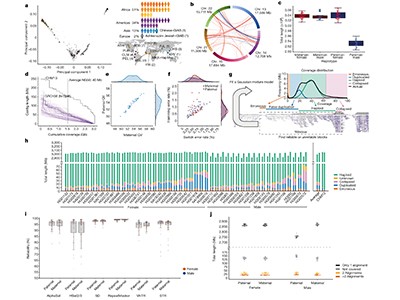
The use of human reference genomes from single individuals is problematic because it introduces biases in how sequences from other human genomes are interpreted. For instance, sequences from other genomes are first commonly aligned to the reference (read mapping) and then reduced to a set of differences from that reference (variant calling). Both processes might yield different results if a different person’s DNA had been used to generate the original reference. This is particularly true for highly diverse and structurally complex regions of the genome. Furthermore, there are hundreds of megabases of DNA that cannot be captured in a reference based on a single genome, because they exist in only a subset of humans4,5.
A pangenome representing many genomes from different ancestries could overcome these issues. However, constructing a pangenome is a complex task. Breakthroughs in the past decade in long-read sequencing technology and computational methods have now enabled this vision to be realized.
Liao and colleagues first generated 94 genome assemblies from 47 individuals (one for each of the two sets of chromosomes that each individual carries). The individuals represent diverse ancestries from around the globe. The assembled genomes, which were generated using a combination of long-read and other sequencing technologies, are highly accurate and nearly complete, and include 119 million base pairs of sequence not included in the draft human reference genome.
The authors used three graph-building methods to construct pangenomes from these assemblies. One of these methods aligns all sequences simultaneously; the others use one genome as a reference and align each subsequent sequence iteratively. The result is a set of publicly available pangenome graphs, along with a rich ecosystem of open-source tools and standardized file formats that researchers can use in a similar way to a linear reference genome.
Liao et al. demonstrated that using their pangenomes for read mapping and variant calling resulted in 34% fewer errors in calling small variants (those shorter than 50 bases) than did using a linear reference. The difference was particularly pronounced in challenging repetitive DNA regions. Impressively, the pangenomes enabled the authors to identify twice as many large genomic alterations, called structural variants, per person than is possible using a linear reference (Fig. 1).

Figure 1 | A human pangenome. The genomes of 47 people of diverse ancestries have been used to generate a draft human pangenome reference3,6,7. Two whole-genome sequences were generated from each individual (one for each of their two sets of chromosomes). The 94 sequences were aligned to form a pangenome graph, which is conceptually similar to an underground-railway map. Boxed regions indicate sequences present in one or more genomes that at a given site, with branching paths indicating sequence variation. The graphs reveal large genome alterations called structural variants, and enable easy analysis of how they vary between individuals.
The human pangenome reference represents a milestone in human genetics. However, challenges remain. Alignment of sequences against highly variable repetitive regions in the pangenome could be improved by more-accurate assemblies or new algorithms. More samples from diverse groups are also needed. Finally, widespread adoption of the pangenome by scientists could take time, because new methods supporting pangenome analysis are continually being developed, and scientists will often require training to use them.
Continued improvements in methods for building and using pangenomes will enable researchers to overcome these challenges. Use of pangenomes has the potential to transform human genomics. This will ultimately make it easier to discover genetic variants that mediate physical and clinical traits and — it is to be hoped — will eventually lead to better health outcomes for many people.
BRIAN MCSTAY & HÁKON JÓNSSON: Untangling repeated sequences
Repetitive DNA regions are challenging to sequence, because it is hard to place them accurately in a genome assembly. These regions include segmental duplications (in which sequences more than one kilobase long are repeated elsewhere in the genome) and the short arms (p-arms) of a subset of chromosomes, dubbed acrocentric chromosomes. Two studies in Nature now use Liao and colleagues’ pangenomes to systematically explore these regions — Guarracino et al.6 to analyse acrocentric p-arms, and Vollger et al.7 to investigate segmental duplications. Their work provides a glimpse of the insights that can be gained from a pangenome reference.
Read the paper: Recombination between heterologous human acrocentric chromosomes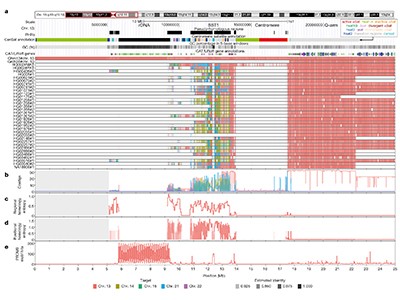
The acrocentric chromosomes (chr13, chr14, chr15, chr21 and chr22 in humans) are those in which the p-arm is considerably shorter than the other (q) arm. Acrocentric p-arms are dedicated to one task: forming sites called nucleoli, where the cell’s protein-assembling machines are made8. P-arms contain nucleolar organizer regions (which encode the RNAs that drive nucleolar formation), highly repetitive DNA and many other shared sequences. This shared, repetitive DNA reflects a phenomenon called heterologous recombination, whereby different acrocentric p-arms pair and cross over to exchange DNA during the cell divisions that generate sperm and eggs. By contrast, in most chromosomes, pairing and crossover are restricted to two copies of the same chromosome (homologous recombination).
In XY sex chromosomes, which also exhibit heterologous recombination, pairing is aided by short regions of homology (near-identical sequences) shared between X and Y. Guarracino et al. constructed a variation graph for acrocentric p-arms using Liao and colleagues’ sequences, and found that they contain pseudo-homologous regions (PHRs). Each PHR is a patchwork of sequence blocks that — as the authors discovered when they compared their graphs with T2T-CHM13 — often show more similarity to the other four acrocentric p-arms in T2T-CHM13 than to the T2T-CHM13 version of themselves. Presumably, these blocks assist heterologous recombination, ensuring that p-arms evolve in concert to preserve their shared role in nucleolar formation.
Robertsonian translocations (ROBs) are phenomena, usually occurring during egg-cell production, whereby the q-arms of two acrocentric chromosomes fuse and most of the p-arms are lost8. Guarracino et al. identified sequences in PHRs at which the breaks that lead to ROBs occur — indicating that ROBs are collateral damage arising from heterologous recombination. Given that ROBs occur in one in 800 human births, we surmise that heterologous recombination between acrocentric chromosomes is both ongoing and frequent. We expect that, as more genomes are added to the pangenome reference, it will be possible to quantify the frequency of this recombination.
Read the paper: Increased mutation and gene conversion within human segmental duplications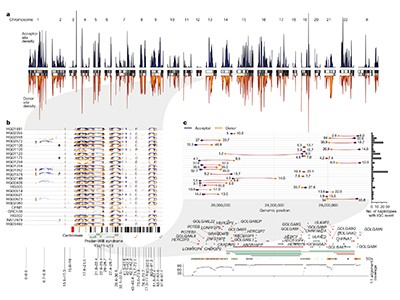
Vollger et al. used the reference to systematically compare variation in segmental duplications with that in non-repetitive parts of the genome (Fig. 1). They found 60% higher sequence diversity in segmental duplications, and showed that these duplications are highly divergent between populations and individuals.
Genes in segmental duplications are susceptible to interlocus gene conversion (IGC) — an exchange of short DNA sequences between non-homologous parts of the duplicated region. Vollger and colleagues identified IGC events by looking for signs of sequence shuffling in the pangenomes, and concluded that these events are probably one of the main reasons that segmental duplications are so diverse. They found that 799 genes had protein-coding regions affected by an IGC.
It is exciting to see accurate characterization of segmental duplications, because duplicated sequences can fuel the evolution of new, specialized roles for a gene. Vollger et al. assessed sequence ‘constraint’ in duplicated genes, with a particular interest in those duplicated during evolution of the human lineage. Constraint is a measure of sequence variability, with less variation indicating that mutations are detrimental to the organism’s viability. Thirty-eight genes were constrained, including members of the NOTCH2 gene family, which has been linked to human-specific changes in brain size during evolution9. The repetitive nature of segmental duplications had previously led to difficulties in assessing constraint for at least 40% of the analysed genes. The authors also found that 171 genes were duplicated and relocated intact to new genomic regions, potentially meaning that their regulation would be rewired. In future, the pangenome project should enable researchers to assess constraint in recently duplicated genes in more depth.
Together, these papers provide a taster of how the human pangenome reference can be used. They reveal how sequence exchanges between repetitive regions of our genome contribute to variation in the population and to our evolution. As the scope of the reference expands, we look forward to further insights into these fascinating genomic regions.
 Read the paper: A draft human pangenome reference
Read the paper: A draft human pangenome reference
 Read the paper: Recombination between heterologous human acrocentric chromosomes
Read the paper: Recombination between heterologous human acrocentric chromosomes
 Read the paper: Increased mutation and gene conversion within human segmental duplications
Read the paper: Increased mutation and gene conversion within human segmental duplications
 The first complete human genome
The first complete human genome
 An assembly line for an improved human reference genome
An assembly line for an improved human reference genome



