- NEWS AND VIEWS
Tumours use a metabolic twist to make lipids
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 566, 333-334 (2019)
doi: https://doi.org/10.1038/d41586-019-00352-1
References
Shevchenko, A. & Simons, K. Nature Rev. Mol. Cell Biol. 11, 593–598 (2010).
Röhrig, F. & Schulze, A. Nature Rev. Cancer 16, 732–749 (2016).
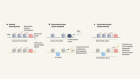
Vriens, K. et al. Nature 566, 403–406 (2019).
Ackerman, D. & Simon, M. C. Trends Cell Biol. 24, 472–478 (2014).
Ge, L., Gordon, J. S., Hsuan, C., Stenn, K. & Prouty, S. M. J. Invest. Dermatol. 120, 707–714 (2003).
Zhang, J. Y., Kothapalli, K. S. D. & Brenna, J. T. Curr. Opin. Clin. Nutr. Metab. Care 19, 103–110 (2016).
Park, H. G. et al. Biochim. Biophys. Acta 1861, 91–97 (2016).
Zelenay, S. et al. Cell 162, 1257–1270 (2015).
Kamphorst, J. J. et al. Proc. Natl Acad. Sci. USA 110, 8882–8887 (2013).
 Read the paper: Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity
Read the paper: Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity
 Histidine metabolism boosts cancer therapy
Histidine metabolism boosts cancer therapy
 Friendly neighbours feed tumour cells
Friendly neighbours feed tumour cells