- NEWS AND VIEWS
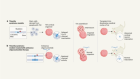
Increased synthesis of a coenzyme linked to longevity can combat disease
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 563, 332-333 (2018)
doi: https://doi.org/10.1038/d41586-018-07088-4
References
Katsyuba, E. et al. Nature 563, 354–359 (2018).
Krehl, W. A., Teply, L. J., Sarma, P. S. & Elvehjem, C. A. Science 101, 489–490 (1945).
Shi, H. et al. N. Engl. J. Med. 377, 544–552 (2017).
Fukoka, S.-I. et al. J. Biol. Chem. 277, 35162–35167 (2002).
Mouchiroud, L. et al. Cell 154, 430–441 (2013)
Gomes, A. P. et al. Cell 155, 1624–1638 (2013).
Pucci, L., Perozzi, S., Cimadamore, F., Orsomando, G. & Raffaelli, N. FEBS J. 274, 827–840 (2007).
Liu, L. et al. Cell Metab. 27, 1067–1080 (2018).
Tran, M. T. et al. Nature 531, 528–532 (2016).
Gariani, K. et al. Hepatology 63, 1190–1204 (2016).
Poyan Mehr, A. et al. Nature Med. 24, 1351–1359 (2018).
Lin, S.-J., Defossez, P.-A. & Guarente, L. Science 289, 2126–2128 (2000).
Williams, P. A. et al. Science 355, 756–760 (2017).
Wang, G. et al. Cell 158, 1324–1334 (2014).
Cantó, C. et al. Nature 458, 1056–1060 (2009).
 Read the paper: De novo NAD+ synthesis enhances mitochondrial function and improves health
Read the paper: De novo NAD+ synthesis enhances mitochondrial function and improves health
 Targeting a fat-accumulation gene
Targeting a fat-accumulation gene
 Tools to eliminate senescent cells
Tools to eliminate senescent cells