
Abstract
FOXP3-expressing regulatory T (Treg) cells, which suppress aberrant immune response against self-antigens, also suppress anti-tumor immune response. Infiltration of a large number of Treg cells into tumor tissues is often associated with poor prognosis. There is accumulating evidence that the removal of Treg cells is able to evoke and enhance anti-tumor immune response. However, systemic depletion of Treg cells may concurrently elicit deleterious autoimmunity. One strategy for evoking effective tumor immunity without autoimmunity is to specifically target terminally differentiated effector Treg cells rather than all FOXP3+ T cells, because effector Treg cells are the predominant cell type in tumor tissues. Various cell surface molecules, including chemokine receptors such as CCR4, that are specifically expressed by effector Treg cells can be the candidates for depleting effector Treg cells by specific cell-depleting monoclonal antibodies. In addition, other immunological characteristics of effector Treg cells, such as their high expression of CTLA-4, active proliferation, and apoptosis-prone tendency, can be exploited to control specifically their functions. For example, anti-CTLA-4 antibody may kill effector Treg cells or attenuate their suppressive activity. It is hoped that combination of Treg-cell targeting (e.g., by reducing Treg cells or attenuating their suppressive activity in tumor tissues) with the activation of tumor-specific effector T cells (e.g., by cancer vaccine or immune checkpoint blockade) will make the current cancer immunotherapy more effective.
Similar content being viewed by others

Introduction
A number of studies have shown that self-antigen or tumor antigen-specific CD4+ and CD8+ T cells are present in healthy individuals1,2,3,4. How such self- or tumor-reactive T cells are controlled in healthy or tumor-bearing individuals remains to be determined. Mechanisms for the maintenance of immunological self-tolerance (i.e., unresponsiveness to self-antigens) not only prevent autoimmunity but also hamper effective tumor immunity because many tumor antigens recognized by autologous lymphocytes are normal self-antigens or quasi-self-antigens with genetic mutations. This is one reason why it is difficult to elicit strong tumor immunity in cancer-bearing patients by cancer vaccine alone5. It also suggests that effective tumor immunity can be evoked by breaching a certain mechanism(s) of immunological self-tolerance systemically or locally in tumor tissues.
Among the various mechanisms of immunological self-tolerance, immune suppression by endogenous Foxp3+CD25+CD4+ Treg cells is essential and indispensable as illustrated by spontaneous autoimmune disease development when Treg cells are rendered deficient. For example, mutations of the gene encoding the Treg-specific transcription factor Foxp3 impair Treg cell development and cause a fatal multi-organ autoimmune disease called immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome6. Depletion of Foxp3+CD25+CD4+ Treg cells by a variety of methods is also able to cause similar autoimmune diseases in otherwise normal rodents7.
On the other hand, it is now well substantiated that a large number of Treg cells infiltrate into tumor tissues of various cancers and their abundant presence is often associated with poor clinical prognosis. Experimentally, the role of Treg cells in tumor immunity was first demonstrated by an attempt to determine a common basis between tumor immunity and autoimmunity8. Removal of Treg cells using cell-depleting anti-CD25 antibody, either by in vivo antibody administration to mice or transfer of cell suspension depleted in vitro of CD25+ Treg cells into histocompatible T-cell-deficient mice, effectively eradicated a variety of inoculated syngeneic tumors8,9. The mice showed an increase of tumor-infiltrating CD8+ T cells with strong tumor-specific killing activity, and upon re-challenge with the same tumor cells, exhibited more rapid rejection than the primary rejection, indicating the establishment of tumor-specific immunity8,10. These studies have thus demonstrated that the removal of Treg cells is able to evoke effective anti-tumor immunity by abrogating immunological unresponsiveness to syngeneic tumors, albeit it may also cause autoimmunity, especially if Treg cells are depleted systemically.
In this review, we discuss molecular basis of Treg functions and their behavior in tumor tissues, and strategies to target Treg cells, in particular their subsets, in order to evoke effective anti-tumor immunity in humans, without eliciting deleterious autoimmunity.
Treg cell function in relation to tumor immunity
T-cell receptor repertoire of Treg cells
The T-cell receptor (TCR) repertoire of Treg cells is broad and skewed to a certain extent to recognizing self-antigens. That is, in the course of T-cell selection in the thymus, a developing Treg cell exhibits a higher TCR affinity than a conventional T (Tconv) cell for the MHC/self-peptide ligand that selects both11. Assuming that TCR recognition of peptides is cross-reactive (and degenerate) and a particular TCR is able to recognize a million different peptides of 10 amino acid length12,13, the TCR repertoire of Treg cells as well as Tconv cells is broad and able to recognize a wide spectrum of self and non-self antigens including quasi-self-tumor antigens. Given the antigen-primed state of endogenous Treg cells (as illustrated by higher level expression of T-cell accessory molecules such as LFA-1), it is reasonable to assume that Treg cells recognizing a particular self- or tumor antigen are more easily activated than naive Tconv cells recognizing the same antigen, ensuring Treg-mediated dominant tolerance14.
Treg-mediated suppression mechanisms
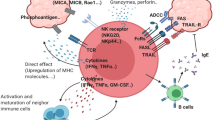
Treg cells are able to control not only T cells but also B cells, NK cells, dendritic cells (DCs), and macrophages via humoral and cell-cell contact mechanisms6. A variety of molecules are involved in Treg-mediated suppression mechanisms, including CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), IL-2, IL-10, TGF-β, IL-35, GITR (glucocorticoid-induced TNF receptor), LAG3 (lymphocyte-activation gene 3), granzyme B, adenosine, and cAMP6 (Figure 1 and Table 1). Given that ectopic Foxp3 expression in Tconv cells is able to confer Treg-like suppressive activity, the molecule(s) mediating a core suppressive mechanism may well be controlled by Foxp3. In addition, among various mechanisms of Treg-dependent suppression, those important for maintaining self-tolerance (i.e., the suppression mechanisms whose impairment causes autoimmune disease) have the most impact on tumor immunity. On these assumptions, there are only a few molecules whose expression is controlled by Foxp3 directly or indirectly and whose deficiency abrogates Treg-suppressive function and causes severe autoimmune diseases. The candidates include IL-2, IL-2 receptor subunits, and CTLA-4. Foxp3 indeed controls the expression of these molecules and deficiencies of IL-2, CD25 (IL-2 receptor α-chain), CD122 (IL-2 receptor β-chain), or CTLA-4 produce similar autoimmune diseases as observed in Foxp3 deficiency6.

Treg suppression mechanisms. Treg cells, which scarcely produce IL-2, deprive IL-2 from the surrounding via their high affinity IL-2 receptor, making it unavailable for responder T cells. They also constitutively express CTLA-4, which down-modulates CD80/CD86 expression by antigen-presenting cells (APCs), thus depriving co-stimulatory signal to responder T cells. Treg cells also produce immune-suppressive cytokines such as IL-10, which also down-modulates APC functions. Under this deprivation of co-stimulatory signal, responder T cells with high-affinity TCRs for the presented antigen die by apoptosis, those with intermediate affinity TCRs are rendered anergic, and those with low-affinity TCRs stay dormant. This IL-2/IL-2 receptor-dependent and CTLA-4-dependent mechanism forms a core basis of Treg-mediated suppression in various tissues including cancer.
A cardinal feature of Treg cells is that they constitutively express high-affinity IL-2 receptor composed of CD25, CD122, and CD132 (common γ-chain) at a high level, but scarcely produce IL-2. Treg cells therefore highly depend on exogenous IL-2, which is mainly produced by activated Tconv cells, for their survival and proliferation. Foxp3 binds to and attenuate AML1 and NFAT, two transcription factors necessary for IL-2 production, thus repressesing IL-2 production15,16. In fact, neutralization of circulating IL-2 by administration of anti-IL-2 antibody can compromise Treg function and survival, causing severe autoimmunity as produced by Treg deficiency6. On the other hand, the constitutive expression of high-affinity IL-2 receptor combined with self-insufficiency in IL-2 production determines that Treg cells absorb IL-2 from their surroundings, thus limiting the amount of IL-2 available for Tconv cells and consequently suppressing the activation and proliferation of the latter17. Accordingly, addition of exogenous IL-2 abrogates Treg-suppressive function in vitro14,18. Therefore, IL-2 and IL-2 receptor can be key targets for controlling Treg cell survival and suppressive function19.
CTLA-4 is a highly potent co-inhibitory molecule expressed constitutively by Treg cells and by Tconv cells after activation. In mice, Treg-specific deletion of CTLA-4 elicits systemic hyper-proliferation of Tconv cells and fatal autoimmune diseases affecting multiple organs, including severe myocarditis20. Recently, heterozygous CTLA-4 mutations in humans were identified in patients with multiple autoimmune symptoms accompanied by impaired suppressive function of Treg cells21,22. As CTLA-4 has much higher affinity than CD28 for their common ligands CD80 and CD86, CTLA-4 expressed by Treg cells may physically outcompete CD28 on Tconv cells for binding to CD80/CD86 on antigen-presenting cells, thereby inhibiting co-stimulation of Tconv cells23,24. Furthermore, CTLA-4 on Treg cells down-modulates expression of CD80 and CD86 on DCs, thereby hindering the activation of Tconv cells at this level20,25,26. Thus, Treg expression of CTLA-4 is essential for Treg-mediated immune suppression.
Treg cells require TCR stimulation to exert suppressive activity; without antigen stimulation, they fail to suppress immune response14. TCR stimulation of Treg cells further upregulates CTLA-4 and other accessory molecules, particularly adhesion molecules such as LFA-1, whose deficiency compromises suppressive activity. When TCR affinity for a stimulating antigen is the same between Treg cells and Tconv cells, Treg cells can be activated to exert suppression at a much lower antigen concentration than Tconv cells14. This indicates that constitutively high expression of LFA-1 and other accessory molecules prior to TCR stimulation may contribute to setting a lower threshold for TCR-induced activation of Treg cells27. On the other hand, another key feature of Treg cells is their hypoproliferation upon TCR stimulation in vitro; albeit a fraction of Treg cells can proliferate actively in response to TCR stimulation in vivo14,28,29. An excessive amount of IL-2 or agonistic anti-CD28 antibody is able to abrogate the in vitro hypo-responsiveness. Ectopic expression of Foxp3 in CD4+ T cells also converts them into a state of hypo-responsiveness upon TCR stimulation30. It has also been recently demonstrated that Treg cells, especially Ki-67+ Treg cells, are highly dependent on tonic TCR signaling for their proliferation31,32, and ablation of TCR signaling results in caspase-mediated apoptosis of these cells in mice. It is also of note that Foxp3-dependent repression of the expression of proximal TCR signaling molecules (e.g., ZAP-70) in Treg cells may contribute to their unique sensitivity to TCR stimulation. For example, whereas TCR stimulation upregulates the expression of ZAP-70 in Tconv cells, it downregulates ZAP-70 expression in Treg cells33. Foxp3 binds to the promoter region of ZAP-70 gene33,34,35 and retroviral expression of Foxp3 in Tconv cells reduces their ZAP-70 expression33. It remains to be determined how TCR signaling attenuation at the level of ZAP-70 may contribute to Treg-specific functions. It might affect Treg cell selection in the thymus to skew the TCR repertoire toward higher self-reactivity11. As another possibility, the TCR signal attenuation might rescue Treg cells from activation-induced cell death upon antigen exposure, enabling them to survive better than Tconv cells and exert dominant control over the latter at tumor sites.
In summary, constitutively high expression of CD25 and CTLA-4, dependency on exogenous IL-2, and TCR stimulation have essential roles in producing and shaping Treg functions, especially Treg-mediated suppression. Indeed, adapting this triad is able to convert Tconv cells into Treg-like suppressive T cells effective in vivo and in vitro, indicating these three events are minimally required for constructing Treg-suppressive activity26. These molecular processes are also good targets to control Treg function and development in the context of tumor immunity.
Treg migration to nonlymphoid tissues
Treg cells are widely distributed in both lymphoid and nonlymphoid tissues, including tumors, where they affect immune responses and inflammation. Treg cells in nonlymphoid tissues are predominantly effector Treg cells (CD44hi CD62Llo) and are highly proliferative36. In mice, migration of Treg cells to nonlymphoid tissues, such as the skin and the lungs, requires chemokine receptor CCR4 (C-C chemokine receptor type 4)37. Treg cells deficient in CCR4 expression fail to migrate, and this causes severe inflammatory diseases in the skin and the lungs.
Cell fate and function of Treg-suppressed Tconv cells
Until recently, the fate of responder T cells after they are suppressed by Treg cells was poorly understood (e.g., if they die by apoptosis, become anergic, or stay dormant during the period of suppression). A recent in vitro study has now shown that the presence of a large number of Treg cells can render self-antigen-/tumor antigen-specific human CD8+ T cells (e.g., recognizing Melan-A antigen) anergic38. These anergic CD8+ T cells are hyporesponsive to antigen stimulation (i.e., hypoproliferative and producing very little IL-2 and other cytokines), express co-inhibitory molecules such as CTLA-4, and are phenotypically naive (i.e., CD45RAhigh and CCR7+). Induction of anergy depends on the intensity of Treg suppression (determined by the number and suppressive activity of the Treg cells) and TCR affinity of the responder T cells. Upon Treg-mediated down-modulation of CD80/CD86 expression by antigen-presenting cells, as discussed above, T cells with high affinity TCR for the stimulating antigen die by apoptosis, whereas T cells with intermediate affinity TCR are rendered anergic, and T cells with low-affinity TCR stay dormant. Thus, Treg cells can induce long-term self-tolerance, and hinder effective tumor immunity, by determining the fate of self-reactive or tumor-reactive T cells (Figure 1).
It is of note that a sizable fraction of self-antigen-specific CD8+ T cells (e.g., those specific for Melan-A as detected by peptide/MHC tetramer staining) present in the peripheral blood of normal individuals are CTLA-4-expressing but naive in phenotype (i.e., CCR7+ and CD45RAhigh) and functionally anergic. In addition, CTLA-4-negative naive CD8+ T cells positive for the same tetramer staining can be activated to proliferate in response to antigen stimulation. It appears that the latter population of non-anergic naive T cells give rise to effector T cells to cause autoimmune disease or mediate effective tumor immunity when Treg-mediated suppression is breached, as discussed below.
Functional/phenotypic heterogeneity of human FOXP3+ T cells
There is accumulating evidence that FOXP3+ T cells in humans are heterogeneous in phenotype and function, consisting of suppressive and non-suppressive subpopulations. For example, naive CD4+ T cells transiently express FOXP3 at a low level upon in vitro TCR stimulation; yet they are not measurably suppressive. Attempts to delineate suppressive and non-suppressive FOXP3+CD4+ T cells present in the peripheral blood of healthy individuals have shown that FOXP3+CD4+ T cells can be divided into three subpopulations based on expression levels of FOXP3 (or CD25) and the cell surface molecule CD45RA39 (Figure 2). They are: (i) FOXP3loCD45RA+CD25lo cells (Fraction I or Fr. I), designated as naive or resting Treg cells; (ii) FOXP3hiCD45RA−CD25hi cells (Fr. II), designated as effector or activated Treg cells, which are terminally differentiated from Fr. I cells upon antigen stimulation and highly suppressive; and (iii) FOXP3loCD45RA−CD25lo non-Treg cells (Fr. III), which do not possess suppressive activity but can secrete pro-inflammatory cytokines. Furthermore, adjacent populations of highly suppressive Fr. II and non-suppressive Fr. III can be better differentiated by the expression of CD15s (sialyl Lewis x), a sugar antigen, on suppressive Treg cells, at least for those in the peripheral blood40. This classification of FOXP3+CD4+ T cells is instrumental in defining suppressive or non-suppressive FOXP3+ subpopulations, delineating developmental stages of Treg cells, and assessing their adaptive processes in physiological and pathological immune responses.

Functional classification of human FOXP3+CD4+ T-cell subpopulations in tumor tissue. Human FOXP3+ T cells in the peripheral blood and lymph nodes are composed of heterogeneous subpopulations containing suppressive Treg cells (naive and effector Treg cells) and activated non-Treg cells without suppression function. These subpopulations are designated as Fraction (Fr.) I, II, and III for naive Treg (nTreg), effector Treg (eTreg), and non-Treg cells, respectively. CD25 surface marker can be used in the place of FOXP3 because of their correlative expression in humans. Majority of cancers are infiltrated predominantly by effector Treg cells, whereas certain cancers are infiltrated by both effector Treg cells and non-Treg cells. Tumor-infiltrating effector Treg cells predominantly express various cell surface molecules including CTLA-4, CCR4, and PD-1.
Tumor-infiltrating Treg cells
Accumulating studies have demonstrated that a large number of Treg cells infiltrate into various types of tumors in humans and mice. In humans, tumors in the head and neck41, breast42, lung43, liver44, gastrointestinal tract45,46, pancreas47, and ovary48,49 have been shown to harbor a large number of tumor-infiltrating Treg cells50. Importantly, decreased ratios of tumor-infiltrating CD8+ T cells to FOXP3+ Treg cells were shown to correlate with poor prognosis, especially in patients with breast51, gastric46, and ovarian cancer48,49. Furthermore, a recent meta-analysis of previously published data indicated that in majority of solid tumors in the cervix, kidney, breast, and melanomas, high frequency of tumor-infiltrating FOXP3+ cells was significantly negatively correlated with patients' survival52.
In contrast, prognosis of patients with Hodgkin lymphoma or colorectal cancer (CRC) containing increased numbers of Treg cells has been controversial53,54,55,56,57. Some studies have indicated that tumor infiltration of FOXP3+ T cells is correlated with a better prognosis, whereas others have shown the contrary. These contradictory results may arise from an improper interpretation of the heterogeneous FOXP3+ cells (i.e., functional Treg cells and non-Treg cells as described above) as a single population of Treg cells. Using the function-based scheme to classify FOXP3+ cells infiltrating the CRC tissues, our recent study has shown that CRC patients can be separated into two groups: one group with tumors infiltrated predominantly by suppression-competent effector Treg cells and the other group with tumors infiltrated with a sizable fraction of FOXP3+ non-Treg cells in addition to effector Treg cells58. In the latter group, FOXP3+ non-Treg cells secrete inflammatory cytokines and the cytokine production is correlated with expression of TGF-β and IL-12 genes in the tumor tissue. In this group, high FOXP3 gene expression shows significantly better prognosis than low FOXP3 gene expression. In contrast, in the group with tumors infiltrated predominantly by effector Treg cells, high FOXP3 gene transcription indicates poor prognosis compared with low FOXP3 transcription. Thus, it is critically important to assess heterogeneity of FOXP3+ T cells in the tumor tissues in order to evaluate their contribution to anti-tumor immune response in various cancers (Figure 2).
Cancer immunotherapy targeting Treg cells
Recent progress in cancer immunotherapy targeting Treg cells, either deliberately or inadvertently, suggests that molecules relatively specific to Treg cells are good candidates for Treg depletion or functional modulation. These molecules include CTLA-4, GITR, CCR4, PD-1, OX-40, and LAG3, as well as aforementioned CD25 and CD15s (Figure 2).
Checkpoint blockade antibody with possible Treg-depleting effects
One of the recent breakthroughs in cancer immunotherapy is the clinical use of anti-CTLA-4 antibody, often referred to as the checkpoint blockade therapy. Two fully humanized monoclonal antibodies against CTLA-4 (Ipilimumab and Tremelimumab) have been tested in patients with various cancer types, including melanoma, prostate cancer, and renal cell carcinomas. The results from a large, randomized, multi-center phase III clinical study for Ipilimumab showed a significant advantage in metastatic melanoma patients59,60. Yet, how Ipilimumab augments anti-tumor immunity is still unclear. CTLA-4, as discussed above, is constitutively expressed on Treg cells, whereas it is transiently expressed by conventional T cells upon activation. Although anti-CTLA-4 mAb was first suggested to augment the activity of tumor-infiltrating CD8+ and CD4+ T cells, recent studies have suggested another possibility that anti-CTLA-4 mAb predominantly affects Treg cells, thereby enhancing anti-tumor immune responses. Using Fc receptor-deficient mice, the anti-tumor activity of anti-CTLA-4 mAb was shown to be dependent on antibody-dependent cellular cytotoxicity of Treg cells in tumor tissues instead of affecting re-activation of Tconv cells61,62,63. In cancer patients, strong correlations have been reported between the clinical efficacy of Ipilimumab and reduction of Treg cell numbers in tumor tissues64,65. It is likely that the Treg-depleting effect of anti-CTLA-4 mAb targets effector Treg cells, which are abundant in tumor tissues and express high levels of CTLA-4. It remains to be determined whether anti-PD-1 antibody (Nivolumab), another checkpoint blockade antibody, possesses a Treg-depleting effect in tumor tissues.
Depletion of effector Treg cells in tumor tissues
As discussed above, in various cancers effector Treg cells are the most abundant cell type among FOXP3+ T cells in the tumor tissues. In order to distinguish and selectively deplete tumor-infiltrating Treg cells while preserving other Treg cells critical for suppressing autoimmunity, one strategy is to specifically target effector Treg cells, which are highly activated, proliferative, and prone to death by apoptosis66. As effector Treg cells are the predominant population in tumor tissues, depleting effector Treg cells would shift the balance in tumor microenvironment from immune suppression to immune activation against tumor cells, despite a continual supply of effector Treg cells from intact naive Treg cells. For this purpose, surface molecules expressed specifically or selectively on effector Treg cells are good targets. For example, CCR4 is predominantly expressed by effector Treg cells, not by naive Treg cells and Th2 cells which do not contribute significantly to tumor immunity regulation in humans67, and Treg migration and infiltration into various tumor tissues appear to be dependent on the expression of CCR4 ligands (i.e., CCL17 and CCL22) produced by tumor cells or infiltrating macrophages48,68. Indeed, the use of anti-CCR4 antibody has been shown to be effective in depleting effector Treg cells selectively and augmenting the induction of tumor antigen-specific CD4+ and CD8+ T cells in vivo67.
Agonistic antibody affecting Treg suppression
GITR is another molecule that is expressed by Treg cells and can serve as a target for functional modulation. Treg cells constitutively express high level of GITR compared with Tconv cells; however, upon activation, Tconv cells also express GITR, whereas the level of GITR on Treg cells is further upregulated. In mice, administration of agonistic anti-GITR antibody (non-depleting) can abrogate Treg cell-mediated suppressive function and enhance the effector function of Tconv cells to break down immunological self-tolerance69,70,71. The same antibody in tumor-bearing mice can indeed break immunological self-tolerance and evoke potent anti-tumor immune response with an increase in IFN-γ-producing CD8+ and CD4+ T cells, which can eradicate established tumors72. Furthermore, a chimeric anti-GITR antibody generated by replacing the Fc portion of the original rat IgG2b with a murine IgG2a has shown effective tumoricidal activity by selectively depleting Treg cells in mice61. The agonistic antibody for GITR is under clinical trials to test its efficacy in melanoma and other advanced solid tumor patients. Agonistic antibodies specific for other TNFR super family molecules, such as OX40, are under clinical investigation66.
Small molecules for Treg depletion or functional modulation
In addition to antibody-mediated Treg depletion therapy, small molecule drugs targeting characteristic features of Treg cells have been shown to be effective. One example is metronomic administration of cyclophosphamide, an anti-neoplastic used frequently in traditional chemotherapy. As cyclophosphamide is an alkylating agent that interferes with DNA replication, it kills highly proliferating cells. Administration of high-dose cyclophosphamide severely affects all T cells. However, when used at low doses over a long term, the drug has been shown to selectively reduce highly proliferating Treg cells including those in the tumor tissues, and enhance anti-tumor immune responses in humans and rodents73,74,75,76. Furthermore, an increasing number of studies have shown the potentials and efficacy of low-dose metronomic cyclophosphamide in combination with other immunotherapeutic agents77,78.
Another potential strategy to augment tumor immunity via controlling Treg cells is to target TCR signaling molecules, which are differentially controlled in Treg and Tconv cells. As discussed above, ZAP-70 is specifically repressed in Treg cells upon TCR activation33. Targeting ZAP-70 may therefore enable selective reduction of TCR signaling by interfering with TCR proximal signaling molecules, resulting in selective death of Treg cells, in particular effector Treg cells, due to signal deprivation-induced apoptosis (Tanaka et al., unpublished). Similarly, an inactivating mutation (D910A mutation) of phosphatidylinositol-3-kinase (PI3K) p110δ or conditional knockout of PI3K in Treg cells in mice effectively augmented anti-tumor immune responses without incurring autoimmunity in the mutant mice79. Although these findings need to be confirmed in humans, these studies suggest that TCR-proximal signaling molecules can be good targets for developing small molecule compounds to achieve selective depletion of tumor-infiltrating effector Treg cells.
Autoimmunity and Treg-targeting cancer immunotherapy
Cancer immunotherapies such as CTLA-4 and PD-1 blockade are frequently accompanied by serious autoimmunity59,80,81. In general, the effectiveness of anti-tumor responses tends to correlate with the development of autoimmune diseases82,83, especially when a systemic Treg cell depletion approach is adopted. There are, however, several ways to evoke effective tumor immunity without causing serious autoimmune reaction by targeting specific populations of Treg cells. One is to selectively target effector Treg cells in tumor tissues, thus preserving the naive Treg cells in other tissues needed to prevent autoimmunity. The second strategy aims to adjust the degree and duration of Treg depletion in order to promote tumor immunity while curbing autoimmune responses, taking advantage of the observation that induction of autoimmune disease generally needs more thorough and durable Treg cell depletion than induction of anti-tumor immune responses. An additional approach to control tumor-infiltrating Treg cells is direct injection of Treg-depleting antibodies to tumor tissues. It is important to note the efficacy of Treg-targeting anti-tumor immunotherapy can be assessed by monitoring Treg cell numbers and function in the blood. Assessing the genetic makeup (e.g., HLA haplotypes) of the patients is also important to determine the susceptibility to autoimmunity due to Treg cell reduction.
Conclusions
Since the discovery of Treg cells as a key mediator of immunological self-tolerance, a common immunological basis for Treg-mediated suppression of autoimmunity and tumor immunity has been extensively explored. The role of Treg cells in tumor immunity is now widely accepted and therapies targeting Treg cells are under active investigation. Combination of Treg cell attenuation (e.g., by reducing Treg cell numbers or reducing their suppressive activity in tumor tissues) with the activation of tumor-specific effector T cells (e.g., by cancer vaccine or immune checkpoint blockades) may mutually enhance each individual treatment. For the combination to be more effective, the sequence of the treatments is critically important (Figure 3). Tumor killing by anti-cancer drugs or ionizing irradiation, or effective tumor antigen vaccination may release self-antigens and tumor-associated antigens and cause local inflammation; and this may recruit and activate Treg cells in tumor tissues, consequently hampering ensuing anti-tumor immune responses. To strongly activate the effector T cells, it is therefore necessary to deplete Treg cells or attenuate their suppressive activity prior to other immunological treatments such as vaccination and checkpoint blockade. We envisage that the combination of various rationally sequenced immunological interventions will make current cancer immunotherapy more effective in the clinic.

Combination and sequence are the key for effective cancer immunotherapy. See text.
References
Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW . Autoreactive T cells in healthy individuals. J Immunol 2004; 172:5967–5972.
Nishikawa H, Jäger E, Ritter G, Old LJ, Gnjatic S . CD4+ CD25+ regulatory T cells control the induction of antigen-specific CD4+ helper T cell responses in cancer patients. Blood 2005; 106:1008–1011.
Yu W, Jiang N, Ebert PJR, et al. Clonal deletion prunes but does not eliminate self-specific αβ CD8+ T lymphocytes. Immunity 2015; 42:929–941.
Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM . Virus-specific CD4+ memory-phenotype T cells are abundant in unexposed adults. Immunity 2013; 38:373–383.
Rosenberg SA, Yang JC, Restifo NP . Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10:909–915.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M . Regulatory T cells and immune tolerance. Cell 2008; 133:775–787.
Sakaguchi S . Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 2004; 22:531–562.
Shimizu J, Yamazaki S, Sakaguchi S . Induction of tumor immunity by removing CD25+CD4+T cells: a common basis between tumor immunity and autoimmunity. J Immunol 1999; 163:5211–5218.
Onizuka S, Tawara I, Shimizu J, et al. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 1999; 59:3128–3133.
Yamaguchi T, Sakaguchi S . Regulatory T cells in immune surveillance and treatment of cancer. Semin Cancer Biol 2006; 16:115–123.
Morikawa H, Sakaguchi S . Genetic and epigenetic basis of Treg cell development and function: from a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol Rev 2014; 259:192–205.
Wooldridge L, Ekeruche-Makinde J, Van Den Berg HA, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem 2012; 287:1168–1177.
Birnbaum ME, Mendoza JL, Sethi DK, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 2014; 157:1073–1087.
Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol 1998; 10:1969–1980.
Ono M, Yaguchi H, Ohkura N, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 2007; 446:685–689.
Wu Y, Borde M, Heissmeyer V, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006; 126:375–387.
Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ . CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol 2007; 8:1353–1362.
Thornton AM, Shevach EM . CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 1998; 188:287–296.
Boyman O, Sprent J . The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 2012; 12:180–190.
Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008; 322:271–275.
Schubert D, Bode C, Kenefeck R, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20:1410–1416.
Kuehn HS, Ouyang W, Lo B, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014; 345:1623–1627.
Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med 2000; 192:303–310.
Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994; 1:405–413.
Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011; 332:600–603.
Yamaguchi T, Kishi A, Osaki M, et al. Construction of self-recognizing regulatory T cells from conventional T cells by controlling CTLA-4 and IL-2 expression. Proc Natl Acad Sci USA 2013; 110:E2116–E2125.
Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN . Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 2015; 528:225–230.
Itoh M, Takahashi T, Sakaguchi N, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol 1999; 162:5317–5326.
Fisson S, Darrasse-Jeze G, Litvinova E, et al. Continuous activation of autoreactive CD4+CD25+ regulatory T cells in the steady state. J Exp Med 2003; 198:737–746.
Hori S, Nomura T, Sakaguchi S . Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299:1057–1061.
Levine AG, Arvey A, Jin W, et al. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 2014; 15:1–10.
Vahl JC, Drees C, Heger K, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity 2014; 41:722–736.
Ohkura N, Hamaguchi M, Morikawa H, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012; 37:1–15.
Marson A, Kretschmer K, Frampton GM, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 2007; 445:931–5.
Samstein RM, Arvey A, Josefowicz SZ, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 2012; 151:153–166.
Smigiel KS, Richards E, Srivastava S, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 2014; 211:121–136.
Sather BD, Treuting P, Perdue N, et al. Altering the distribution of Foxp3+ regulatory T cells results in tissue-specific inflammatory disease. J Exp Med 2007; 204:1335–1347.
Maeda Y, Nishikawa H, Sugiyama D, et al. Detection of self-reactive CD8+ T cells with an anergic phenotype in healthy individuals. Science 2014; 346:1536–1540.
Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30:899–911.
Miyara M, Chader D, Sage E, et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3 high regulatory T cells in humans. Proc Natl Acad Sci USA 2015; 112:7225–7230.
Schaefer C, Kim GG, Albers A, et al. Characteristics of CD4+CD25+ regulatory T cells in the peripheral circulation of patients with head and neck cancer. Br J Cancer 2005; 92:913–920.
Liyanage UK, Moore TT, Joo H-G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 2002; 169:2756–2761.
Wolf AM, Wolf D, Steurer M, et al. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res 2003; 9:606–612.
Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F . Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res 2005; 65:2457–2464.
Ichihara F, Kono K, Takahashi A, et al. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res 2003; 9:4404–4408.
Sasada T, Kimura M, Yoshida Y, et al. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer 2003; 98:1089–1099.
Hiraoka N, Onozato K, Kosuge T, Hirohashi S . Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 2006; 12:5423–5434.
Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942–949.
Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 2005; 102:18538–18543.
Nishikawa H, Sakaguchi S . Regulatory T cells in tumor immunity. Int J Cancer 2010; 127:759–767.
Bates GJ, Fox SB, Han C, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 2006; 24:5373–5380.
Shang B, Liu Y, Jiang S, Liu Y . Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 2015; 5:15179.
Álvaro T, Lejeune M, Salvadó MT, Banham AH, Roncador G, Montalba C . Outcome in Hodgkin's lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res 2005; 11:1467–1473.
Wilke CM, Wu K, Zhao E, Wang G, Zou W . Prognostic significance of regulatory T cells in tumor. Int J Cancer 2010; 127:748–758.
Salama P, Phillips M, Grieu F, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 2009; 27:186–192.
Frey DM, Droeser RA, Viehl CT, et al. High frequency of tumor-infiltrating FOXP3+ regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer 2010; 126:2635–2643.
Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ . Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology 2009; 137:1270–1279.
Saito T, Nishikawa H, Wada H, et al. Two FOXP3+CD4+ T-cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016; 22:679–684.
Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517–2526.
Bulliard Y, Jolicoeur R, Windman M, et al. Activating Fcγ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 2013; 210:1685–1693.
Simpson TR, Li F, Montalvo-Ortiz W, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 2013; 210:1695–1710.
Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 2013; 1:32–42.
Liakou CI, Kamat A, Tang DN, et al. CTLA-4 blockade increases IFNγ-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA 2008; 105:14987–14992.
Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci USA 2008; 105:3005–3010.
Nishikawa H, Sakaguchi S . Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 2014; 27:1–7.
Sugiyama D, Nishikawa H, Maeda Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA 2013; 110:17945–17950.
Faget J, Biota C, Bachelot T, et al. Early detection of tumor cells by innate immune cells leads to Treg recruitment through CCL22 production by tumor cells. Cancer Res 2011; 71:6143–6152.
Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S . Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 2002; 3:135–142.
McHugh RS, Whitters MJ, Piccirillo CA, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 2002; 16:311–323.
Stephens GL, McHugh RS, Whitters MJ, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol 2004; 173:5008–5020.
Ko K, Yamazaki S, Nakamura K, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med 2005; 202:885–891.
Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 2004; 34:336–344.
Motoyoshi Y, Kaminoda K, Saitoh O, et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep 2006; 16:141–146.
Ghiringhelli F, Menard C, Puig PE, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 2007; 56:641–648.
Ge Y, Domschke C, Stoiber N, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother 2012; 61:353–362.
Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med 2009; 206:1103–1116.
Le DT, Jaffee EM . Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res 2012; 72:3439–3444.
Ali K, Soond DR, Piñeiro R, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014; 509:407–411.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443–2454.
Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016; 54:139–148.
Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA 2003; 100:8372–8377.
Attia P, Phan GQ, Maker AV, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 2005; 23:6043–6053.
Acknowledgements
We would like to thank Dr James B Wing for critical reading of this manuscript. This study was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Sports, and Culture (MEXT), and Japan Agency for Medical Research and Development (AMED). The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tanaka, A., Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res 27, 109–118 (2017). https://doi.org/10.1038/cr.2016.151
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.151
Keywords
This article is cited by
-
Comprehensive analysis of m6A modification in immune infiltration, metabolism and drug resistance in hepatocellular carcinoma
Cancer Cell International (2024)
-
Integrated multi-omic analysis and experiment reveals the role of endoplasmic reticulum stress in lung adenocarcinoma
BMC Medical Genomics (2024)
-
Loss of chromosome Y in regulatory T cells
BMC Genomics (2024)
-
cAMP-PKA/EPAC signaling and cancer: the interplay in tumor microenvironment
Journal of Hematology & Oncology (2024)
-
AMPKα2 promotes tumor immune escape by inducing CD8+ T-cell exhaustion and CD4+ Treg cell formation in liver hepatocellular carcinoma
BMC Cancer (2024)
