
Abstract
The evolution of glucocorticoid drugs was driven by the demand of lowering the unwanted side effects, while keeping the beneficial anti-inflammatory effects. Potency is an important aspect of this evolution as many undesirable side effects are associated with use of high-dose glucocorticoids. The side effects can be minimized by highly potent glucocorticoids that achieve the same treatment effects at lower doses. This demand propelled the continuous development of synthetic glucocorticoids with increased potencies, but the structural basis of their potencies is poorly understood. To determine the mechanisms underlying potency, we solved the X-ray structures of the glucocorticoid receptor (GR) ligand-binding domain (LBD) bound to its endogenous ligand, cortisol, which has relatively low potency, and a highly potent synthetic glucocorticoid, mometasone furoate (MF). The cortisol-bound GR LBD revealed that the flexibility of the C1-C2 single bond in the steroid A ring is primarily responsible for the low affinity of cortisol to GR. In contrast, we demonstrate that the very high potency of MF is achieved by its C-17α furoate group completely filling the ligand-binding pocket, thus providing additional anchor contacts for high-affinity binding. A single amino acid in the ligand-binding pocket, Q642, plays a discriminating role in ligand potency between MF and cortisol. Structure-based design led to synthesis of several novel glucocorticoids with much improved potency and efficacy. Together, these results reveal key structural mechanisms of glucocorticoid potency and provide a rational basis for developing novel highly potent glucocorticoids.
Similar content being viewed by others

Introduction
Glucocorticoids, such as prednisone, dexamethasone (DEX) and budesonide, are the most effective anti-inflammatory drugs. They are widely used to treat inflammation and autoimmune diseases such as asthma, arthritis, lupus, and Crohn's disease1,2. These drugs exert their physiologic roles through binding to the glucocorticoid receptor (GR), a ligand-activated transcription factor of the nuclear receptor superfamily. In the absence of glucocorticoid, GR resides in the cytoplasm and associates with chaperone proteins, such as hsp90 and hsp70. The binding of hormone causes a conformational change in GR, leading to its translocation to the nucleus, where it exerts its transcriptional activity, either activation (transactivation) or repression (transrepression). In transactivation, GR dimerizes, binds directly to a specific glucocorticoid response element, and then recruits coactivators to activate transcription. In transrepression, the general model is that GR binds other transcription factors (e.g., NF-κB and AP-1) to become indirectly tethered to their binding sites through protein-protein interactions. Upon being tethered near a target promoter, GR represses downstream gene expression3. It is generally believed that transrepression does not require GR dimerization4,5.
Transrepression is the major mechanism through which glucocorticoids act as anti-inflammatory agents6. The tethering of GR to the NF-κB/AP-1-driven promoters leads to the transcriptional repression of major downstream proinflammatory factors, including proinflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6), chemokines (e.g., CCL2, CCL19), and enzymes associated with the onset of inflammation (e.g., COX2, MMP13, and phospholipase A2)2. Because of their fast action and sustainable effects, glucocorticoids remain the first choice for treating inflammatory diseases. However, the long-term use of glucocorticoids, especially at high doses, has many adverse consequences, including diabetes, hypertension, obesity, and osteoporosis7,8. Most of these consequences are attributed to the transactivation of GR. For instance, glucocorticoids induce the genes encoding rate-limiting enzymes of the gluconeogenesis pathway, glucose-6-phosphatase and phosphoenol pyruvate carboxykinase, in liver, thus augmenting the de novo synthesis of glucose and eventually leading to weight gain or diabetes9,10. Glucocorticoids also induce a key regulatory gene of bone development, Dickkopf-1 (DKK1), upregulation of which leads to osteoporosis and bone loss11. It is generally observed that many of the side effects of glucocorticoids are associated with use of high-dose glucocorticoids12,13,14. For example, a “threshold pattern” was observed for the use of prednisone: administration at 7.5 mg per day causes glaucoma, depression, and high blood pressure12. These side effects are caused by GR transactivation as well as non-target activation of other receptors, such as mineralocorticoid receptor (MR), whose activation causes high blood pressure15. Thus, it is important to develop highly potent and selective glucocorticoids to reduce the unwanted side effects.
Potency and efficacy are two key pharmacokinetic parameters of glucocorticoids. While efficacy is the maximal activity that a given drug can achieve, usually at maximal concentration, potency is the concentration of the drug required to reach half maximal activity (EC50). For two glucocorticoids that have the same efficacy, a highly potent one will require a lower dose to achieve the same treatment effect14,15. Importantly, a glucocorticoid may have different potencies for transactivation and transrepression; for example, gene induction by GR via DEX requires a 5-6-fold higher glucocorticoid concentration than gene repression16,17,18. This differential response provides an opportunity for developing highly potent glucocorticoids that can be used at low doses to achieve full repression of inflammation signals, while minimizing transactivation activity and side effects. Finally, the development of insensitivity to glucocorticoid therapy (glucocorticoid resistance) is a major problem in treating common inflammatory diseases, such as chronic obstructive pulmonary disease, rheumatoid arthritis, and inflammatory bowel disease19. Glucocorticoid resistance is also an unsolved issue for white blood cell cancers, especially childhood acute leukemia20. Several mechanisms of glucocorticoid resistance have been identified or proposed, including a change of kinase pathways, alteration of cofactors, and loss or mutation of receptors19,21. One common observation is that the potency of ligand for the receptor is decreased in glucocorticoid-resistant patients. Such patients treated with highly potent glucocorticoids have shown improvement, but the effect gradually decreased22. Therefore, there is an urgent need to develop a new generation of more potent glucocorticoids.
Cortisol is an endogenous glucocorticoid produced by the adrenal gland. Cortisol has a relative low potency and receptor binding ability when compared to the most commonly used synthetic glucocorticoids, such as DEX23. On the other hand, mometasone furoate (MF) is a potent glucocorticoid used to treat inflammatory skin disorder (Elocon), asthma (Asmanex), and nasal sinus inflammation (Nasonex)24,25. MF has a lipophilic furoate ester group at the C-17α position of the steroid D ring, which is believed to be the origin of its high potency26. Here, we determined the crystal structures of the GR ligand-binding domain (LBD) bound to MF and cortisol, which revealed the underlying mechanisms that discriminate the ligand potencies of MF and cortisol. We then exploited the observed structure mechanisms to increase potency and efficacy of several newly designed glucocorticoids, which could serve as starting leads for the development of novel therapeutics for the treatment of inflammatory diseases.
Results
Overall structures of the cortisol- and MF-bound GR LBDs
Crystallization of the GR LBD has always been a challenge due to its low solubility. The original GR LBD structure was determined with a high-affinity ligand, DEX, bound to a GR LBD in which F602 was replaced with serine to improve protein solubility27. However, cortisol is a much weaker ligand than DEX and the F602S mutation is not sufficient for stabilization of the GR LBD bound to cortisol, an endogenous hormone (Supplementary information, Figure S2, lane 1). To identify amino acids that might increase GR LBD solubility without affecting the overall structure, we aligned GR with the closest members of the steroid hormone family, MR, androgen receptor (AR), and progesterone receptor (PR), which are much more soluble than GR (Supplementary information, Figure S1A). Besides F602, residues C622, T668, S674, and V675 differ from the conserved sequence of the family; we therefore replaced these amino acids with their conserved counterparts (F602A, C622Y, T668V, S674T, and V675I, termed AYVTI). Most of these residues are found at the inside of the proteins (Supplementary information, Figure S1B), with the PR residues having better packing in the PR LBD structure28. Indeed, the AYVTI GR LBD has much better solubility than the F602S GR LBD when bound to cortisol (Supplementary information, Figure S2, lane 2). The mutated GR LBD can be expressed and purified with a yield > 5 mg/L. However, we were not able to obtain crystals of this mutated GR LBD bound to cortisol or MF. The GR LBD has several lysine and glutamic acid residues in helix H9 and we reasoned that these long side chains may impede crystallization. Introduction of surface entropy reduction mutations (K669A/K703A for MF and E684A/E688A for cortisol) allowed us to obtain receptor-ligand complexes that remained soluble (Supplementary information, Figure S2, lanes 3 and 4) and formed crystals (Supplementary information, Figure S3). These altered residues are far away from the ligand-binding pocket (Supplementary information, Figure S1B) and do not affect ligand-mediated GR transactivation or transrepression functions (Supplementary information, Figure S4).
The overall structures of the MF- and cortisol-bound GR LBDs (Figure 1A) are similar to that of the DEX-bound GR LBD, with 11 helices packing into three layers of a helical sandwich bundle and the ligand-binding cavity buried in the lower part of the bundle (the statistics of data collection and refinement are summarized in Supplementary information, Table S1). The overall architecture of cortisol-bound GR LBD is almost identical to that of DEX-bound GR LBD. In contrast, there are some notable differences between MF-bound GR LBD and DEX-bound GR LBD, including the orientation of the loop N-terminal to helix 1 (labeled “1” in Figure 1B), an expansion of the loop region between helices 5 and 6 (labeled “2” in Figure 1B), and a change in the C-terminal orientation of the AF2 helix (labeled “3” in Figure 1B). The ligand binding modes of cortisol and MF are well defined by clear electron density of the bound ligands and the surrounding pocket residues (Figure 1C).

Overall structures of cortisol-bound GR LBD and MF-bound GR LBD. (A) Architectures of cortisol-bound GR LBD and MF-bound GR LBD. (B) Structural comparison of the DEX-bound GR LBD (green) and the MF-bound GR-LBD (magenta). Arrows indicate differences between them: 1, loop region N-terminal to helix 1; 2, loop region between helices 5 and 6; 3, orientation of the C-terminus of the AF2 helix. (C) Composite omit map (2fo-fc) of cortisol and MF in the ligand-binding pocket of GR LBD contoured to 1σ.
Potencies and affinities of cortisol, DEX and MF
The change of chemical structures (Figure 2A) of cortisol, DEX and MF correlated with the evolution of glucocorticoids from low to high potency. The cortisol structure contains a basic 4-ring steroid backbone, and DEX differs from cortisol by addition of a Δ1 (C1-C2) double bond, a C-16 α-methyl and a C-9 α-fluoro group (Figure 2A, green color). MF in turn differs from DEX by a C-9 chloro instead of fluoro group and is further modified by addition of a C-21 α-chloro group and, more importantly, a lipophilic furoate ester group at C-17α (Figure 2A, magenta color), replacing the hydroxyl group of DEX and cortisol. To systematically compare the efficacy and potency of MF, DEX and cortisol, we determined full dose-response curves for both GR transactivation and transrepression in a cell-based reporter assay. We measured GR transactivation activity using an MMTV-driven luciferase reporter system (Figure 2B). MF and DEX showed almost the same efficacy (maximal activity) at the saturation concentration (1 μM), whereas cortisol at its saturation concentration (10 μM) had only 80% of the efficacy of DEX. Relative to DEX, there was a large leftward shift of the MF dose-response curve, indicating that MF is 20-fold more potent than DEX. On the other hand, the cortisol curve had a large rightward shift, showing 10-fold less potency than DEX. The EC50 values for MF, DEX and cortisol in transactivation were 0.33 nM, 6.7 nM and 70 nM, respectively.

Potency of cortisol, DEX, and MF. (A) Chemical structures of cortisol, DEX, and MF. Steroid rings (A-D) are labeled in red, carbon atom numbers in black. Differences between DEX and cortisol are highlighted in green in the DEX structure. The furoate group of MF is labeled in magenta. (B, C) Dose-response curves for cortisol, DEX and MF on induction of MMTV-Luc reporter gene activity and repression of AP1-Luc reporter gene activity in AD293 cells. RLU, Relative Luciferase Units. Error bars = SD, n = 3. (D) In vitro GR radioligand binding assay. [3H]-DEX bound to GR was competed with unlabeled MF, DEX or cortisol. cpm, counts per minute. Error bars = SD, n = 2.
To study GR transrepression, an AP1-driven luciferase reporter was used (Figure 2C). MF, DEX and cortisol showed similar efficacies at their saturation concentrations. Again, MF showed much higher (60-fold) potency than DEX, and cortisol was much weaker than DEX; the EC50 values for MF, DEX and cortisol in transrepression were 0.005 nM, 0.32 nM and 2.7 nM, respectively. Consistent with the frequent observation that induction requires a higher steroid concentration, the induction potency was at least 10-fold lower than the repression potency for each compound. This difference provides an opportunity to dissociate transactivation from transrepression via the use of very low doses of glucocorticoid. For example, at 0.1 nM, MF reaches 95% of transrepression efficacy but only 25% of transactivation efficacy (Figure 2B and 2C).
Generally, high potency is determined by a high affinity for the receptor, but cellular cofactors also play important roles29,30. To test the affinity of MF for GR, we performed in vitro GR ligand binding competition assays for MF, DEX and cortisol (Figure 2D), which showed that the order of GR binding affinity was MF > DEX > cortisol. The Ki values for MF, DEX and cortisol were 0.7 nM, 8 nM and 91 nM, respectively. This result was consistent with our result for potency. However, the difference in in vitro determined IC50 values between MF and DEX was only about 10-fold, while the difference in potency was much more, 20-fold for induction and 60-fold for repression (Figure 2B and 2C). The other component of the difference in potency must be contributed by interactions with cellular factors that recognize the surface conformational changes caused by the binding of different ligands.
The flexibility of the C1-C2 single bond contributes to the low affinity of cortisol to GR
To understand the structural mechanism underlying the low affinity of cortisol, we first compared the structures of cortisol-bound GR LBD and DEX-bound GR LBD. Since the overall structure of cortisol-bound GR LBD is essentially the same as that of DEX-bound GR LBD without notable conformation change, functional differences must be due to details in ligand binding. As mentioned above, DEX differs from cortisol only by the C1-C2 double bond, C-9α halogenation, and C-16α methylation (Figure 2A, green color). The C1-C2 double bond of DEX causes the steroid A ring and the C-3-ketone group to become planar, thus allowing the C-3-ketone to readily interact with R611 and Q570 (Figure 3A). In contrast, because of the flexibility of the cortisol C1-C2 single bond, the steroid A ring needs to bend to form a hydrogen bond with R611 and Q570. Also, since the C1-C2 single bond of unbound cortisol oscillates between two conformations (above and below the A-ring plane), a water molecule is required to form a hydrogen bond network to hold the ligand in position. These observations explained the relatively low affinity of cortisol to GR. To confirm the importance of the C1-C2 double bond, we measured the potency of prednisolone, which differs from cortisol only by addition of the C1-C2 double bond (Figure 3B, brown color), in a transactivation assay. Indeed, the C1-C2 double bond of prednisolone caused an about 5-fold leftward shift of the cortisol dose-response curve (Figure 3B) and may therefore account for more than half of the total leftward shift caused by DEX. The remaining increase of potency is likely due to the C-9α halogenation and the C-16 methylation, both of which increase the interaction surface within the receptor pocket (Supplementary information, Figure S5).

The flexibility of the C1-C2 single bond contributes to the low affinity of cortisol. (A) Hydrogen bond network of cortisol (red) and DEX (green) in the ligand-binding pocket of GR LBD. The hydrogen bond distances are marked on the right panels. (B) Dose-response curves of cortisol, prednisolone and DEX for GR transactivation of MMTV-Luc in AD293 cells. Prednisolone differs from cortisol only by having a Δ1 (C1-C2) double bond (brown color in chemical structure). Error bars = SD, n = 3.
The C-17α furoate group is the key determinant of the high affinity of MF
While DEX has an almost flat, two-dimensional structure (Figure 4A), the C-17α furoate ester group of MF sticks out of the ring plane at almost 90°, adding a third dimension to the ligand (Figure 4B). In the DEX-bound GR LBD, there is an empty space above the steroid D ring, a hydrophobic cavity formed by helices 3-7 and the β3-β4 turn (Figure 4C and 4D). In the MF-bound GR LBD structure, the protruding C-17α furoate group expands the ligand-binding pocket slightly and takes up most space of that cavity. The lipophilic C-17α furoate group fits nicely into the hydrophobic cavity and makes extensive hydrophobic interactions with the surrounding F623, I629, M639, and C643 amino acids (Figure 4D), resembling a ball firmly anchored in socket joint and explaining why MF has a 10-fold higher affinity for GR than DEX.

The C-17α furoate group allows MF to fully occupy the GR ligand-binding pocket. (A, B) Three-dimensional structures of DEX (green) and MF (magenta). (C) Structure alignment of DEX and MF in the ligand-binding pocket of GR LBD. The C-17α furoate group of MF expands the GR ligand-binding pocket and fully occupies the hydrophobic cavity above the steroid D ring. The meshes indicate the boundaries of ligand-binding pocket. (D) Detailed hydrophobic interactions of the C-17α furoate group with residues in the hydrophobic cavity of the GR LBD ligand-binding pocket.
Q642 plays a key role in recognizing glucocorticoids of different potencies
The basic recognition of glucocorticoids by the GR LBD has been described27,31,32. As in the DEX-bound GR LBD, Q570 and R611 interact with the C-3 keto group of the steroid A ring, N564 interacts with the C-11 hydroxyl group of the steroid C ring, and T739 interacts with the side chain C-21 carbonyl group (Figure 3A). These four pairs of important hydrogen bonds hold the steroid backbone tightly in position. Relative to DEX-bound and cortisol-bound GR LBDs, the intruding C-17α furoate group of MF causes only one large change inside the ligand-binding pocket, which is the movement of Q642 in helix 7 (Figure 5A). In the DEX-bound GR LBD structure, Q642 is perpendicular to the axis of helix 7 and forms hydrogen bonds with the C-17α hydroxyl group of DEX. Upon binding of MF, the C-17α furoate group pushes Q642 away, bending it nearly 90° into a position parallel to the axis of helix 7 (Figure 5A).

Q642 plays critical roles in recognizing ligands of different potencies. (A) Detailed interaction of Q642 with different ligands; green, DEX-bound GR-LBD; magenta, MF-bound GR-LBD. (B) Transactivation activity of GR Q642 mutant proteins at ligand concentration below saturation (DEX, 10 nM; MF, 1 nM). Error bars = SD, n = 3. (C) Dose-response curves of wild-type (WT; black) and Q642N (red) GR for MMTV-Luc activity in AD293 cells in the presence of MF, DEX or cortisol. Error bars = SD, n = 3.
Since the Q642 orientation is the only large change in the ligand-binding pocket upon binding of MF, we tested the effects of replacing Q642 with either a smaller (Q642A), larger (Q642F), hydrophobic (Q642L), charged (Q642E, Q642K), or just slightly smaller (Q642N) residue. When tested in the presence of either MF or DEX at subsaturating concentrations (MF, 1 nM; DEX, 10 nM), for one mutant protein, Q642N, DEX activity was nearly abolished, while MF activity remained maximal (Figure 5B). Thus, a single mutation could completely separate the activity of MF from that of DEX. Other replacements caused loss of most of the activity for both DEX and MF; the exception, Q642L, retained half of the activity in the presence of MF, but no or very low activity in the presence of DEX. The C-17α furoate group also slightly changed the conformation around M560 and M639, but mutations altering these residues had little effect on relative glucocorticoid selectivity (Supplementary information, Figure S6).
To analyze the prominent role of Q642 in recognizing ligands of different potencies, we determined full dose-response curves for MF, DEX, and cortisol — representing high, medium, and low potency, respectively — in binding to Q642N in a GR transactivation assay (Figure 5C). For MF, the dose-response curve of Q642N was indistinguishable from that of the wild type. For DEX, Q642N caused a large rightward shift of the curve relative to wild type, with the EC50 changing from 7.5 nM to 40 nM, a 5-fold decrease in potency, and for cortisol, Q642N was inactive, even at saturation concentration. Thus, the single mutation Q642N has the ability to completely separate ligands of high, medium, and low potency. When binding a medium- or low-potency glucocorticoid (e.g., DEX or cortisol), Q642 forms a hydrogen bond with the C-17α hydroxyl group to tether the bound ligand in position within the ligand-binding pocket. When binding to the highly potent ligand MF, Q642 is pushed away by the C-17α lipophilic group. This change, coordinated with other small changes caused by ligand binding, perturbs helices 5, 6, and 7, leading to the expansion of the loop between helix 5 and helix 6 and changing the orientation of the C-terminus of the AF2 helix (Figure 1B), which results in the characteristics of high potency.
To investigate the exact role of Q642 in binding of different ligands, we tested the ligand binding ability of GR Q642A, for which DEX has almost no transactivation activity at a single unsaturated concentration (Figure 5B). In an in vitro binding assay using the cytosol from AD293 cells that expressed either wild-type GR or GR Q642A, the Q642A mutant showed a substantial loss of binding affinity to DEX compared to wild-type GR (Kd (Q642A) = 22.3 nM vs Kd (WT) = 5.2 nM), but still retained some affinity at high ligand concentration (Supplementary information, Figure S7A). On the other hand, Q642A showed almost no transactivation activity, even at a saturating concentration of DEX, in a reporter assay (Supplementary information, Figure S7B). These data show that the lack of DEX transactivation of GR Q642A is due to both a decrease in ligand affinity and a conformation change that inhibits GR activation. Unlike with DEX and cortisol, Q642 does not form a hydrogen bond with MF. To determine whether Q642A still has the ability to bind MF, we performed a competition binding experiment using the GR Q642A mutant protein (Supplementary information, Figure S7C). Both MF and cortisol were able to compete the binding of 3H-DEX to GR Q642A, but with a large decrease of affinity (Ki for MF and cortisol were 9 nM and 250 nM, respectively, compared to that of wild type GR at 0.7 nM and 91 nM, respectively). Taken together, these results suggest that Q642 acts as a pillar to support the ridge of helix 7 by forming a hydrogen bond with C-17α hydroxyl group-containing ligands, while pushing away ligands with a C-17α furoate group. Substituting Q642 with a small residue like alanine may lead the ridge of helix 7 to collapse and thus to lose all transactivation.
Molecular design of novel glucocorticoids
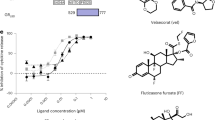
The MF-bound GR LBD structure revealed that the high potency of MF is achieved primarily by the affinity-enhancing interactions of the C-17α furoate group with ligand-binding pocket residues. We applied these structural insights to the design of novel glucocorticoids with the aim to increase their potencies. We had previously determined the crystal structure of the GR LBD bound to deacylcortivazol (DAC)32, a synthetic glucocorticoid that has a bulky phenylpyrazole group replacing the C-3 ketone of the steroid A ring (Figure 6A). DAC is a high-affinity glucocorticoid that has been used in treating glucocorticoid-resistant childhood acute leukemia22. However, DAC has strong cytotoxicity when used at high concentration, and cancer cells gradually develop resistance to the drug33. Since DAC has a hydroxyl group at the C-17α position, the MF-bound GR LBD structure suggested the possibility of developing a “super-potent” glucocorticoid by replacing the C-17α hydroxyl group of DAC with a C-17α furoate group. To avoid the strong side effects of DAC, we used the DAC moiety VSG24 as a starting point (Figure 6A). VSG24 itself has almost no activity in the MMTV-Luc reporter assay. Replacement of the C-17α hydroxyl group with a furoate ester group (VSG22) greatly increased both potency and efficacy, resulting in an EC50 change by > 1 000-fold. The potency of VSG22 was better than that of DEX or DAC (Figure 6A). We also see a similarly great improvement of potency (> 1 000-fold) of VSG22 compared to VSG24 in transrepression assay using an AP1-Luc reporter in AD293 cells (Supplementary information, Figure S8A). These data demonstrate the power of a lipophilic group at the C-17α position to increase glucocorticoid potency. The dramatic change of potency via a simple change at C-17α indicates the power of this strategy to enhance glucocorticoid potency. A similar strategy has been used to enhance the potency of fluticasone propionate, resulting in the highly potent compound fluticasone furoate, a widely used asthma medication34.

Introduction of a C-17α furoate group increases the potencies of glucocorticoid compounds. (A) Dose-response curves of VSG22, VSG24, DAC and DEX on MMTV-Luc activity in AD293 cells. The furoate group in the chemical structures is highlighted in magenta. Error bars = SD, n = 3. (B) A side-by-side comparison of compounds with and without introduced C-17α furoate group (magenta) on potency of transactivation activity on MMTV-Luc in AD293 cells. Error bars = SD, n = 3.
The DAC-bound GR LBD structure had revealed a previously undiscovered channel opened up by the phenylpyrazole group of DAC32. We designed and synthesized several derivatives by introducing an even larger group at the C-3 ketone (VSG02, VSG03, VSG15; Figure 5B). All these compounds showed relatively low potency for GR. Based on the MF-bound GR LBD structure, we introduced a furoate group at the C-17α position, producing the corresponding compounds VSG10, VSG11, and VSG14, respectively. For all three compounds, the introduction of C-17α furoate group greatly increased both GR potency and efficacy of both induction and repression (Figure 6B and Supplementary information, Figure S8B), further suggesting that adding the C-17α furoate group could be a general strategy to augment the potency of designed glucocorticoids.
Discussion
Glucocorticoids have been in use for almost 60 years and they remain the first choice for treating many inflammatory and autoimmune diseases. However, long-term use of glucocorticoids can cause many adverse effects, which are further accentuated by high doses of glucocorticoids. Understanding the structural basis of GR transactivation and transrepression is key to developing novel glucocorticoids with reduced side effects. However, the low expression level of GR in bacterial systems, especially for low-affinity ligands, has hampered the structural study of this important cellular regulator. By comparing conserved residues within the steroid receptor family, we could successfully identify amino acid replacements that could facilitate receptor expression without affecting receptor function. This strategy may allow the structural study of GR bound to important low-affinity ligands, such as non-steroidal agonists, which hold great potential as next generation glucocorticoids.
The development of highly potent glucocorticoids was driven by two urgencies: reducing the side effects of glucocorticoids caused by high-dose usage, and countering clinical glucocorticoid resistance. While ligand affinity is a determining factor in potency, it is not the only one. Cellular cofactors also play crucial roles by recognizing surface differences caused by ligand binding, and subtle variations induced by binding of different ligands can have profound effects on cofactor selectivity. Different strategies had been applied to modify the rigid cortisol backbone to increase potency, and led to the development of DEX. A structural comparison of cortisol-bound GR LBD and DEX-bound GR LBD shows that among the modifications, the Δ1 double bond is critical for optimally positioning the C3 ketone, which forms a key hydrogen bond with R611. Subsequently, researchers found that a lipophilic ester group, such as an alkyl or propionate ester26, at the C-17α position can strongly enhance glucocorticoid activity. One of the most commonly used asthma drugs, fluticasone propionate (FP), was generated by replacing the hydroxyl group with a propionate ester at the C-17α position. These data suggested the presence of a hydrophobic cavity above the steroid D ring in the ligand-binding pocket. Further optimization of FP with a furoate ester group to replace the propionate ester created a highly potent glucocorticoid, fluticasone furoate (FF), indicating that the furoate group might fit best in the cavity.
Although the structure of the FF-bound GR LBD had been solved35, the structural mechanism of the high potency of MF was not defined. Here, we revealed that the high potency of MF is attributable to both the C-17α furoate group occupying the full ligand-binding pocket and the surface conformation changes caused by ligand binding. Using mutagenesis, we demonstrated that a single amino acid residue, Q642, plays a crucial role in recognizing the C-17α furoate group and coordinating the positioning of other amino acid side chains. Q642N differs from the wild-type protein by only one methyl group, yet is sufficient to completely separate the activities of MF, DEX, and cortisol, indicating how precisely the receptor activity is regulated.
We have demonstrated that the C-17α furoate group can serve as an “anchor” point to position low-affinity ligands precisely and firmly in the ligand-binding pocket. The success in modifying DAC derivatives designed for increased dissociating properties demonstrates the power of this strategy for designing therapeutic dissociated glucocorticoids, glucocorticoids with reduced clinical glucocorticoid resistance symptom, or non-steroid glucocorticoid compounds (those compounds generally show poor affinity to receptor). In summary, we have solved the first crystal structure of the GR LBD bound to a physiological ligand, the low-potency glucocorticoid cortisol, as well as the structure of the LBD bound to the clinically important high-potency synthetic ligand MF. In combination with biochemical and mutational analysis, we have structurally identified the critical determinants of glucocorticoid affinity and potency, and validated these determinants through structure-based design and synthesis of highly potent glucocorticoids.
Materials and Methods
Protein expression and purification
The GR LBD (residues 525-777) containing mutations F602A, C622Y, T668V, S674T, V675I, K699A, and K703A for complexation with MF and mutations F602A, C622Y, T668V, S674T, V675I, E684A, and E688A for complexation with cortisol were expressed as a 6× His-GST fusion protein from the expression vector pET24a (Novagen). The modified fusion proteins contain a His6-tag (MKKGHHHHHHG) at the N-terminus and a thrombin protease site between GST and the GR LBD. BL21(DE3) cells transformed with the expression plasmids were grown in LB broth at 16 °C to an OD600 of ∼1 and induced with 0.1 mM IPTG and 50 μM MF or cortisol. Cells were harvested, resuspended in 200 ml extract buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2 M urea, 10% glycerol, and 1 μM ligand) per 12 liters of cells, and passed three times through a French Press with pressure set at 1 000 Pa. The lysate was centrifuged at 20 000 rpm for 30 min, and the supernatant was loaded on a 25 ml nickel column. The column was washed with 700 ml extract buffer and eluted with 300 ml of 50% Buffer B (25 mM Tris, pH 8.0, 500 mM imidazole, 10% glycerol, 1 μM ligand). The GR LBD was cleaved overnight with thrombin at a protease/protein ratio of 1:1 000 in the cold room while being dialyzed against 20 mM Tris, pH 8.0, 500 mM NaCl, 10% glycerol, 1 μM ligand. The H6GST tag was removed by binding to a Ni-NTA nickel column. The flow-through was further purified by gel filtration (20 mM Tris, pH 8.0, 500 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol, 1 μM ligand). The MF-bound protein was complexed with SRC2-3 peptide (SPKKKENALLRYLLDKDDTKD) and filter-concentrated to 6 mg/ml. The cortisol-bound GR LBD was complexed with a shorter version of the SRC2-3 peptide (KENALLRYLLDKDD) and 0.2% β-octylglucoside, and filter-concentrated to 7 mg/ml.
Crystallization
The MF-GR crystals were grown at room temperature in hanging drops containing 1 μl of the protein complexed with SRC2-3 peptide and 2 μl of the well solution containing 0.1 M sodium citrate (pH 6.0) and 2.2 M sodium chloride. The cortisol-GR crystals were grown at room temperature in hanging drops containing 1 μl of the protein complex and 1 μl of the well solution containing 0.1 M imidazole, pH 6.5, and 1 M sodium acetate trihydrate. 30% sucrose in the well buffer was used as a cryoprotectant for both crystal forms.
Structure determination
The CCP4 program PHASER was used for molecular replacement36, with the GR LBD/DEX structure (PDB code: 1M2Z)27 as the search model. The initial model was manually rebuilt and refined using CNS37 and the CCP4 program REFMAC538. All structure figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger LLC).
Cell transfection and reporter assay
AD293 cells were split at 20 000/well in 24-well plates 1 day before transfection. For transactivation, 100 ng pHHLuc (MMTV-Luc) plasmid, 0.1 ng pRShGR together with 5 ng phRGtkRenilla were transfected by X-tremeGENE 9 (Roche) into AD293 cells per well. For transrepression, 10 ng AP1-Luc, 100 ng pRShGR, and 5 ng phRGtkRenilla were transfected into AD293 cells per well. One day after transfection, cells were treated with either steroids or vehicle for 16 h. Cells were harvested by addition of 1× Passive Lysis Buffer (Promega), and luciferase activity was assayed by the Dual-Glo Luciferase system (Promega). Data were plotted as firefly luciferase activity normalized to Renilla luciferase activity in Relative Luciferase Units (RLU).
In vitro GR ligand binding assay
The in vitro GR binding assay is similar to the one described previously39. Basically,[3H]-Dex at 25 nM was incubated with 5% GR cytosol plus 20 mM sodium molybdate in TAPS buffer (pH 8.8) and the indicated concentrations of unlabeled competitors. Data were plotted as a standard competition curve by GraphPad Prism 5.
Accession codes
The PDB codes for cortisol-bound GR LBD and MF-bound GR LBD are 4P6X and 4P6W, respectively.
References
Barnes PJ . Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998; 94:557–572.
De Bosscher K, Vanden Berghe W, Haegeman G . The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 2003; 24:488–522.
Lefstin JA, Yamamoto KR . Allosteric effects of DNA on transcriptional regulators. Nature 1998; 392:885–888.
Heck S, Kullmann M, Gast A, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J 1994; 13:4087–4095.
Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998; 93:531–541.
Rosen J, Miner JN . The search for safer glucocorticoid receptor ligands. Endocr Rev 2005; 26:452–464.
Schacke H, Docke WD, Asadullah K . Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 2002; 96:23–43.
Stanbury RM, Graham EM . Systemic corticosteroid therapy--side effects and their management. Br J Ophthalmol 1998; 82:704–708.
Nakae J, Kitamura T, Silver DL, Accili D . The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 2001; 108:1359–1367.
Opherk C, Tronche F, Kellendonk C, et al. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol 2004; 18:1346–1353.
Pinzone JJ, Hall BM, Thudi NK, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009; 113:517–525.
Hoes JN, Jacobs JW, Verstappen SM, Bijlsma JW, Van der Heijden GJ . Adverse events of low- to medium-dose oral glucocorticoids in inflammatory diseases: a meta-analysis. Ann Rheum Dis 2009; 68:1833–1838.
Spies CM, Strehl C, van der Goes MC, Bijlsma JWJ, Buttgereit F . Glucocorticoids. Best Pract Res Cl Rh 2011; 25:891–900.
Hoes JN, Jacobs JW, Buttgereit F, Bijlsma JW . Current view of glucocorticoid co-therapy with DMARDs in rheumatoid arthritis. Nat Rev Rheumatol 2010; 6:693–702.
Frey FJ, Odermatt A, Frey BM . Glucocorticoid-mediated mineralocorticoid receptor activation and hypertension. Curr Opin Nephrol Hypertens 2004; 13:451–458.
Simons SS Jr . What goes on behind closed doors: physiological versus pharmacological steroid hormone actions. Bioessays 2008; 30:744–756.
Wei P, Ahn YI, Housley PR, Alam J, Vedeckis WV . Modulation of hormone-dependent glucocorticoid receptor function using a tetracycline-regulated expression system. J Steroid Biochem Mol Biol 1998; 64:1–12.
Adcock IM, Nasuhara Y, Stevens DA, Barnes PJ . Ligand-induced differentiation of glucocorticoid receptor (GR) trans-repression and transactivation: preferential targetting of NF-kappaB and lack of I-kappaB involvement. Br J Pharmacol 1999; 127:1003–1011.
Barnes PJ, Adcock IM . Glucocorticoid resistance in inflammatory diseases. Lancet 2009; 373:1905–1917.
Kaspers GJ, Pieters R, Klumper E, De Waal FC, Veerman AJ . Glucocorticoid resistance in childhood leukemia. Leuk Lymphoma 1994; 13:187–201.
Haarman EG, Kaspers GJ, Veerman AJ . Glucocorticoid resistance in childhood leukaemia: mechanisms and modulation. Brit J Haematol 2003; 120:919–929.
Gaynon PS, Carrel AL . Glucocorticosteroid therapy in childhood acute lymphoblastic leukemia. Adv Exp Med Biol 1999; 457:593–605.
Baxter JD . Glucocorticoid hormone action. Pharmacol Ther B 1976; 2:605–669.
Onrust SV, Lamb HM . Mometasone furoate. A review of its intranasal use in allergic rhinitis. Drugs 1998; 56:725–745.
McCormack PL, Plosker GL . Inhaled mometasone furoate: A review of its use in persistent asthma in adults and adolescents. Drugs 2006; 66:1151–1168.
Crim C, Pierre LN, Daley-Yates PT . A review of the pharmacology and pharmacokinetics of inhaled fluticasone propionate and mometasone furoate. Clin Ther 2001; 23:1339–1354.
Bledsoe RK, Montana VG, Stanley TB, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002; 110:93–105.
Williams SP, Sigler PB . Atomic structure of progesterone complexed with its receptor. Nature 1998; 393:392–396.
Simons SS Jr . The importance of being varied in steroid receptor transactivation. Trends Pharmacol Sci 2003; 24:253–259.
Simons SS Jr . How much is enough? Modulation of dose-response curve for steroid receptor-regulated gene expression by changing concentrations of transcription factor. Curr Top Med Chem 2006; 6:271–285.
Kauppi B, Jakob C, Farnegardh M, et al. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem 2003; 278:22748–22754.
Suino-Powell K, Xu Y, Zhang C, et al. Doubling the size of the glucocorticoid receptor ligand binding pocket by deacylcortivazol. Mol Cell Biol 2008; 28:1915–1923.
Harmon JM, Schmidt TJ, Thompson EB . Non-glucocorticoid receptor-mediated effects of the potent glucocorticoid deacylcortivazol. Cancer Res 1982; 42:2110–2114.
Valotis A, Hogger P . Human receptor kinetics and lung tissue retention of the enhanced-affinity glucocorticoid fluticasone furoate. Respir Res 2007; 8:54.
Biggadike K, Bledsoe RK, Hassell AM, et al. X-ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor-ligand binding domain. J Med Chem 2008; 51:3349–3352.
Bailey S . The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50:760–763.
Brünger AT, Adams PD, Clore GM, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1998; 54:905–921.
Murshudov GN, Vagin AA, Dodson EJ . Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 1997; 53:240–255.
He Y, Xu Y, Zhang C, et al. Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci Signal 2011; 4:ra44.
Acknowledgements
We thank staff members of the Life Science Collaborative Access Team of the Advanced Photon Source (APS) for assistance with data collection at the beam lines of sector 21, which is in part funded by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (085P1000817). Use of APS was supported by the Office of Science of the US Department of Energy, under contract No. DE-AC02-06CH11357. This study was supported by NIDDK/NIH fund (DK066202 and DK071662), American Asthma Foundation fund (2010), Amway (China), the National Natural Science Foundation of China (91217311), the Chinese Postdoctoral Science Foundation (2012M511158 and 2013T60477), and the Jay and Betty Van Andel Foundation.
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Protein sequence alignment of the LBDs of GR, PR, MR, and AR (PDF 462 kb)
Supplementary information, Figure S2
Protein expression and purification of selected GR LBD mutant proteins (PDF 123 kb)
Supplementary information, Figure S3
Protein crystals and diffraction images of cortisol- and MF- bound GR LBD (PDF 302 kb)
Supplementary information, Figure S4
The transcriptional activities of GR AYVTI mutant proteins (PDF 96 kb)
Supplementary information, Figure S5
Detailed structural comparisons of the steroid C-9α group and C-16 group of Dex and cortisol in the ligand binding pocket of the GR LBD (PDF 300 kb)
Supplementary information, Figure S6
The F623 and M639 mutations cannot separate the transcriptional activities of MF- and DEX-bound GR (PDF 213 kb)
Supplementary information, Figure S7
In vitro binding assay of GR Q642A mutant protein (PDF 309 kb)
Supplementary information, Figure S8
Transrepression properties of compounds with a C-17α furoate group (PDF 344 kb)
Supplementary information, Table S1
Statistics of data sets and structure refinement (PDF 188 kb)
Rights and permissions
About this article

Cite this article
He, Y., Yi, W., Suino-Powell, K. et al. Structures and mechanism for the design of highly potent glucocorticoids. Cell Res 24, 713–726 (2014). https://doi.org/10.1038/cr.2014.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2014.52
Keywords
This article is cited by
-
Corticosteroids in ophthalmology: drug delivery innovations, pharmacology, clinical applications, and future perspectives
Drug Delivery and Translational Research (2021)
-
Preparation and Evaluation of Cubosomes/Cubosomal Gels for Ocular Delivery of Beclomethasone Dipropionate for Management of Uveitis
Pharmaceutical Research (2020)
-
Glucocorticoid receptor control of transcription: precision and plasticity via allostery
Nature Reviews Molecular Cell Biology (2017)
-
Discovery of a highly potent glucocorticoid for asthma treatment
Cell Discovery (2015)
-
The glucocorticoid mometasone furoate is a novel FXR ligand that decreases inflammatory but not metabolic gene expression
Scientific Reports (2015)
