
Abstract
Coronary arteries bring blood flow to the heart muscle. Understanding the developmental program of the coronary arteries provides insights into the treatment of coronary artery diseases. Multiple sources have been described as contributing to coronary arteries including the proepicardium, sinus venosus (SV), and endocardium. However, the developmental origins of coronary vessels are still under intense study. We have produced a new genetic tool for studying coronary development, an AplnCreER mouse line, which expresses an inducible Cre recombinase specifically in developing coronary vessels. Quantitative analysis of coronary development and timed induction of AplnCreER fate tracing showed that the progenies of subepicardial endothelial cells (ECs) both invade the compact myocardium to form coronary arteries and remain on the surface to produce veins. We found that these subepicardial ECs are the major sources of intramyocardial coronary vessels in the developing heart. In vitro explant assays indicate that the majority of these subepicardial ECs arise from endocardium of the SV and atrium, but not from ventricular endocardium. Clonal analysis of Apln-positive cells indicates that a single subepicardial EC contributes equally to both coronary arteries and veins. Collectively, these data suggested that subepicardial ECs are the major source of intramyocardial coronary arteries in the ventricle wall, and that coronary arteries and veins have a common origin in the developing heart.
Similar content being viewed by others

Introduction
Coronary heart disease is the leading cause of death worldwide resulting from pathology of the coronary arteries, the vessels that supply blood to the heart muscle1. Despite their importance to human health, the origins of coronary arteries and their developmental pathways have not been fully delineated. A more complete understanding of the origin and developmental program of coronary vessels would contribute important information to the field of cardiovascular regenerative medicine2.
Scientists have been interested in coronary artery development for many years. Histological observations made a century ago gave rise to the belief that coronary arteries sprout out from the aorta3. With the advent of immunohistochemistry enabling researchers to directly follow vascular cells, it was determined that a vasculature forms first, and then invades the aorta, excluding the possibility of aortic sprouting4. These data were consistent with subsequent avian transplantation studies showing that coronary vascular cells arise from the (pro)epicardium and migrate anteriorly from the posterior side of the heart5,6. This model of coronary artery development became dominant in the field and was thought to be similar in other species.
A recent burst of research based on mouse genetic lineage-tracing techniques has suggested that, in this species, cell types other than the (pro)epicardium contribute to the coronary vasculature. First, mouse lines that express Cre recombinase in the (pro)epicardium induce genetic labeling of either a subset of (Scx-Cre, Sema3d-Cre)7 or very few to no (Wt1-Cre, Tbx18-Cre)8,9,10 coronary endothelial cells (ECs). Other studies have shown that coronary vessels in the embryo express the sprouting marker, Apelin (Apln), and are formed by vascular sprouting from the sinus venosus (SV) and endocardium11,12. These observations are consistent with reports that the SV is physically connected to coronary veins13,14. However, the tracing of SV-derived sprouts into intramyocardial arteries and capillaries in addition to veins suggested that venous endothelium could undergo a reprogramming event to form all coronary vessel subtypes11. Marker analysis suggested that this event involved endothelial dedifferentiation into progenitors that redifferentiate into arteries, capillaries, and veins based on their final location within the heart. In rodents, these locations are such that coronary arteries and capillaries are within the myocardium, whereas veins are on the surface just beneath the epicardium11,15. In spite of these advances, conflicting conclusions emerge on the origin and growth of intramyocardial coronary arteries. For the majority of intramyocardial coronary arteries, recent work suggested SV as origin11, whereas another group concluded that ventricular endocardial cells directly migrate into compact myocardium to form these coronary arteries12.
Here, we address the pattern by which coronary vessels expand through lineage tracing of the derivatives of the early subepicardial ECs. We found that nascent vascular sprouts located just beneath the epicardium give rise to a large proportion of the coronary arteries, capillaries, and veins throughout the ventricle wall at later stages. Clonal analysis of early subepicardial ECs shows that they possess the capability to differentiate into coronary arteries, capillaries, and veins. The distribution of sister cells within the clones reveal that each subepicardial EC equally gives rise to the arterial or venous lineage. These data show that a significant number of ECs within the early subepicardial plexus give rise to cells that migrate into the compact myocardium to form coronary arteries.
Results
Quantification of subepicardial and intramyocardial coronary development
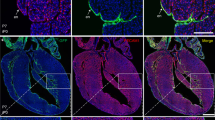
The heart is avascular before E10.5, but is gradually covered by a sprouting vessel network spreading from the SV region over the ventricles of the developing heart (Figure 1A). The earliest coronary vascular plexus is located on the surface just beneath the epicardium, but eventually appears deep within the myocardium where the vessels differentiate into coronary arteries and capillaries (Figure 1B). We carried out a systematic quantification of coronary vessels in the subepicardial (in contact with the epicardium) and intramyocardial (in contact with cardiomyocytes) locations over developmental time. Our data indicated that the ventricle wall contains only subepicardial coronary ECs before E12.5 (Figure 1B). Intramyocardial vessels appear only after E12.5 when they dramatically increase and, by E15.5, outnumber the subepicardial fraction by more than five-fold (Figure 1C). Next, we counted the number of intramyocardial vessels that are in continuity with and within 20 μm of subepicardial ECs. The same criteria were used to assess intramyocardial coronary vessels association with endocardial ECs. This analysis revealed that, as intramyocardial coronary vessels appeared in the ventricle wall, a significant number is continuous with the subepicardial fraction (Figure 1D). In contrast, a consistent, 20-μm wide vascular-free zone that almost never contained ECs separated intramyocardial vessels from the endocardium in the ventricle wall (Figure 1D). The avascular zone separating the endocardium from intramyocardial vessels does not appear to be generated by localized compact myocardial expansion. This is because the whole region (from epicardium to endocardium) is expanding through cardiomyocyte proliferation, and, during this time, intramyocardial and subepicardial vessels continously connect (Figure 1D). In summary, the presence of subepicardial coronary ECs precedes intramyocardial vessel development, and the two are closely associated while a vessel-free border exists around the endocardium throughout all stages (E11.5-E14.5).

Subepicardial and intramyocardial vessels in developing heart. (A) Coronary vessel coverage over the surface of the heart from E11.5 to E14.5 (n = 9-13). Graph calculates area encircled by dotted lines. (B) PECAM staining of E10.5 to E15.5 hearts showing that subepicardial coronary vessels (arrows) develop before intramyocardial vessels (arrowheads). Right panels are boxed regions and results are representative of 5-10 hearts per time point. (C) Quantification of the number of PECAM+ subepicardial and intramyocardial coronary ECs in the left ventricle from E10.5 to E15.5. (D) Continuity analysis by quantifying the number of PECAM+ cells within 20 μm of endocardial (Endo.) or subepicardial (Subepi.) ECs at E11.5 to E14.5. *P < 0.05; n = 6-10. White bar = 0.5 mm; black bar = 200 μm; yellow bar = 50 μm.
Production and characterization of an AplnCreER transgenic mouse line
The spatiotemporal appearance of subepicardial and intramyocardial populations, and the physical association between the two led us to investigate whether a substantial proportion of intramyocardial vessels arise from subepicardial ECs. Here we use the term subepicardial ECs to describe the ECs of a primary subepicardial vascular plexus, which is assessed by their lack of differentiation markers11, and ability to become either artery or vein11. To specifically label the subepicardial fraction and all its progeny, we generated a mouse line expressing an inducible Cre recombinase coupled to the estrogen receptor under the control of the Apln promoter (AplnCreERT2/+) (Supplementary information, Figure S1). In situ hybridization revealed that Apln was expressed in a pattern consistent with that of coronary vessel distribution (Figure 2A). qRT-PCR on sorted Tie2-positive cardiac ECs showed that Apln expression increased from E11.5, whereas pan expression of EC markers Pecam and Vegfr2 (in ratio to Gapdh) was similar across all ages (Figure 2B–2E). Immunostaining for the estrogen receptor (ESR) as a surrogate for Apln expression in the AplnCreERT2/+ line indicated no colocalization with WT1+, TBX18+, or RALDH2+ (pro)epicardial cells from E9.5 to E12.5 (Figure 2F–2J).

Apln and AplnCreERT2 expression map in early developing heart. (A) Whole mount in situ hybridization shows no Apln expression in PEO at E9.5 and Apln expression in a coronary vessel-specific pattern at E11.5 and E12.5. (B) qRT-PCR analysis of heart ventricles shows that pan expression of endothelial markers Pecam and Vegfr2 is similar in different stages, whereas Apln expression begins at E11.5 and subsequently increases. *P < 0.05. n = 4-10. (C) ECs were purified by FACS of dissociated Tie2-GFP hearts. GFP+ and GFP− populations were processed for qRT-PCR. Compared with the pan EC markers Vegfr2 and Pecam, expression of Apln was significantly increased in ECs from E11.5 to E14.5. n = 4-9. (D-E) E12.5 hearts were dissected into either the entire ventricle (V) or apex only (A) for qRT-PCR analysis. At E12.5, Apln expression level was significantly lower in the apex compared with the ventricle, suggesting that most Apln+ sprouting coronary ECs have not reached the apex. NS, not significant; *P < 0.05; n = 8-10. (F-J) CreERT2 (ESR, yellow arrowheads) is not expressed in TBX18+, WT1+, or RALDH2+ (pro)epicardial cells (white arrowheads) or within heart (h) from E9.5 to E12.5. White asterisks indicate atrioventricular groove. (K) Apln-LacZ expression is restricted to the surface of ventricle wall at E12.5 (white arrowheads). Black line outlines the epicardium. PEO, proepicardium; SV, sinus venosus; V, ventricle; A, atrium; LV, left ventricle; RV, right ventricle; Li, liver. Black bar = 0.5 mm; white bar = 100 μm.
Sema3d is expressed in a subset of Wt1−;Tbx18− proepicardial cells that can contribute to coronary ECs7. To test whether Apln is expressed in Sema3d+ proepicardial cell compartment, we generated a Sema3dLacZ/+ mouse line (Supplementary information, Figure S2) and crossed it to AplnCreERT2/+ (Supplementary information, Figure S1). X-gal staining verified that LacZ expression recapitulated the endogenous expression pattern of Sema3d in the proepicardium and epicardium (Supplementary information, Figure S3)7. By immunostaining for ESR on X-gal-treated AplnCreERT2/+;Sema3dLacZ/+ tissue, we found no colocalization of Apln with Sema3d+ proepicardial cells (E9.5, Supplementary information, Figure S4) or Sema3d+ epicardial cells (E10.5 and E11.5, data not shown). Taken together, Apln is not expressed in WT1+, TBX18+, RALDH2+, and SEMA3D+ (pro)epicardial cells in the developing heart from E9.5 to E12.5.
In contrast to the absence of epicardial colocalization, a robust Apln signal was detected in subepicardial coronary ECs and ECs in other vessels of the embryo. Apln was not expressed in cells deep within the heart where the endocardium is located (Figures 2H–2J and 3), which is consistent with previously reported results11,16. A separate Apln reporter line, AplnLacZ/+, verified that Apln expression is restricted to subepicardial ECs, and not in endocardial ECs, at E12.5 (Figure 2K and Supplementary information, Figure S5). Thus, AplnCreER is specific for coronary vascular ECs.

Apln expression map at E9.5 to E12.5. (A-D) Apln is expressed in PECAM+ vascular ECs, but not in endocardial ECs. Asterisks indicate AV groove; red arrow indicates Apln+PECAM+ vascular ECs. PA, pharyngeal arch; Li, liver; h, heart; PEO, proepicardial organ. Representative of three hearts for each time point. White bar = 100 μm.
We next used AplnCreERT2/+ hearts in a functional culture assay to test whether fate-traced cells arise from the SV/atria or ventricle endocardium. Pairing the SV/atria portion of transgenic hearts with wild-type (WT) ventricle gave rise to labeled coronary ECs on the ventricle surface only in the presence of tamoxifen. As a control, the above pairing did not lead to labeled coronary ECs if tamoxifen was not added (Figure 4A). Conversely, these same ECs sprouting from the SV was no longer detectable when a WT SV/atria portion was paired with a transgenic AplnCreERT2/+ ventricle (Figure 4A). Thus, AplnCreERT2/+-labeled coronary vessels arise from the endocardium of SV and/or atria, but less likely from ventricle endocardium. We also paired the SV/atria portion from a Tie2-Cre;Rosa26LacZ/+ mouse with a WT ventricle, and the results were similar (Figure 4B).

Subepicardial ECs contribute substantially to intramyocardial coronary vessels. (A) Tissue recombination assays using AplnCreERT2/+;Rosa26RFP/+ transgenic sinus venosus and atria (SV/A) portions, and WT ventricle with epicardium (V/Epi). In the presence of tamoxifen, marked coronary vessels (arrowheads) could be detected. No tamoxifen was used as control. No marked coronary vessels were detected when transgenic V/Epi was paired with WT SV/A. (B) Tie2-Cre;Rosa26LacZ/+ transgenic SV/A was paired with WT V/Epi. Marked coronary vessels (arrowheads) form on the ventricle. Thus, the SV/A is the source of coronary vessels in this assay. (C) Genetic labeling strategy for lineage tracing of subepicardial ECs. (D) Whole-mount view and tissue section of AplnCreERT2/+;Rosa26LacZ/+ hearts stained with X-gal. Embryos were dosed with tamoxifen at E10.5, which restricts cell labeling to the subsequent 24 h. In section views, asterisks indicate endocardial cushion; black arrowheads point to sprouting subepicardial ECs (X-gal+); red lines with double arrows denote the thickness of ventricle wall. Black dotted lines outline the epicardium; red dotted lines indicate the border between the trabecular and compact myocardial layer. (E) Whole-mount view and tissue section of X-gal-stained E15.5 AplnCreERT2/+;Rosa26LacZ/+ hearts without tamoxifen injection. (F) Whole mount and sectional fluorescence of AplnCreERT2/+;Rosa26RFP/+ heart. (G) Example of sections from E15.5 AplnCreERT2/+;Rosa26mTmG/+ hearts used to quantify the percentage of coronary vessels derived from subepicardial ECs in the ventricle wall. Staining of RFP (tdTomato) and GFP (mG) represents lineage marker as Apln-derived cells. PECAM were used as pan endothelial markers. Tamoxifen was injected at E10.5 for F and G. White arrowheads indicate labeled intramyocardial ECs. Representative of 3. White bar = 0.5 μm, yellow bar = 50 μm. LV, left ventricle; RV, right ventricle; VS, ventricular septum.
Maintaining our cultures for 72 h using both AplnCreERT2/+;Rosa26RFP/+ and Tie2-Cre;Rosa26LacZ/+ E11.5 hearts did not produce ventricles that were as vascularized as those from E14.5 embryos. The inability to fully recapitulate the coronary vessel growth is a limitation of our in vitro assay. However, our data, together with previous work11, support a model that a substantial amount of Apln+ cells originate from endocardium of the SV/atria, whereas much fewer are from ventricular endocardium.
Subepicardial ECs invade the myocardium to form coronary arteries
To investigate the distribution of coronary vessels arising from the earliest subepicardial ECs, we specifically labeled E10.5 and E11.5 Apln+ cells by injecting tamoxifen into AplnCreERT2/+;Rosa26LacZ/+ mice at E10.517, or fluorescence reporter lines Rosa26mTmG/+ that express green-fluorescence protein after Cre recombination18, or Rosa26RFP/+ that express red-fluorescence protein tdTomato after Cre recombination19 (Figure 4C). This induces a permanent genetic lesion that is transferred to all daughter cells17,18,19. Analyzing a time course post-injection demonstrated that subepicardial ECs migrate step-wise down from AV groove and into the myocardium to contribute to significant portions of the entire coronary vasculature within the ventricle walls (Figure 4D). Remarkably, hearts injected with a single E10.5 dose of tamoxifen gave rise to a fate-traced pattern that was very similar to that of endogenous expression11, that is, most coronary cells were lineage-labeled. To exclude the potential penetration problem of X-gal whole mount staining, we sectioned AplnCreERT2/+;Rosa26LacZ/+ hearts and performed both the X-gal reaction and β-GAL immunostaining directly on slides. These experiments confirmed that endocardial cells were not genetically labeled, but that a substantial number of intramyocardial coronary vessels were (Supplementary information, Figure S6). Interestingly, although coronary vessels in the compact myocardium of the ventricle wall were mostly derived from subepicardial ECs, very few within the ventricular septum were genetically labeled. This result indicates that septal vessels are not descendants of subepicardial ECs or from coronaries in the ventricle wall (Figure 4D). Again, this is not due to a penetration problem, as X-gal staining on sections verified that septal vessels were β-GAL-negative (data not shown).
To strengthen our findings, we used two additional reporter lines and detected the same pattern when AplnCreERT2/+ was coupled with the Rosa26RFP/+ or Rosa26mTmG/+ reporter alleles18,19 (Figure 4F and 4G). As a control, genetic labeling was dependent on the presence of tamoxifen (Figure 4E, Supplementary information, Figure S7A–S7C). Immunostaining results were also verified by direct detection of fluorescence reporters (Figures 4G, 5 and Supplementary information, Figure S8), and the emission spectra were always consistent with the expected signal range for each fluorophore (Supplementary information, Figure S7D).

Subepicardial ECs are the major sources of intramyocardial coronary vessels in the ventricle wall. Differential labeling of coronary vessels in the ventricular septum (VS) when dosed at E10.5 and E12.5. Labeled cells in the septum only with the E12.5 tamoxifen injection show that the E10.5 dose is inactive by this time. *P < 0.05; NS, not significant; n = 3-4. White bar = 100 μm. LV, left ventricle; RV, right ventricle; VS, ventricular septum.
Tissue sections revealed that labeled subepicardial ECs produced vessels that migrate into the ventricular myocardium in addition to spreading along its surface (Figure 4D, 4F and 4G). Contributions of subepicardial ECs to intramyocardial coronary arteries were high. We quantified this by counting the number of lineage-labeled intramyocardial cells and comparing it to the total number of PECAM+ coronary ECs in the same location. Of 10 samples with high tamoxifen induction efficiency (this is determined by heavy labeling of vessels on the embryonic body surface), the average percentage of labeled coronary vessels was more than 90% and the numbers were the same for both the left and right ventricle (left ventricle wall, 93.21% ± 1.90 %; right ventricle wall, 92.01% ± 2.30 %). In four samples, subepicardial ECs contributed to as high as 99% of the intramyocardial coronary vessels in the ventricle wall at E15.5 (Figure 4G). Thus, subepicardial coronary ECs or subepicardial ECs present at E10.5-E11.5 can give rise to a substantial amount of intramyocardial coronary vessels within the compact myocardium of free ventricle wall.
We performed additional control experiments to provide direct evidence that E10.5 tamoxifen injections only label subepicardial ECs at E10.5-E11.5 and not intramyocardial coronary vessels present after E12.5. We took advantage of the observations described above that an E10.5 tamoxifen injection does not label coronary vessels in the ventricular septum. As septal vessels start to form at E12.5 (data not shown), a tamoxifen injection at this time point should induce their labeling (Figure 5). Indeed, an E12.5 tamoxifen injection labeled both septal and intramyocardial vessels, whereas labeling induced at E10.5 was restricted to the intramyocardial population (P < 0.05, Figure 5). Importantly, both strategies produced high-efficiency labeling of intramyocardial coronary vessels in the ventricle wall (Figure 5). Thus, the E10.5 tamoxifen injection does not induce recombination at E12.5 and only labels subepicardial ECs, providing a basis for specific genetic lineage tracing of this population at E10.5-E11.5, and detection of their progeny at E12.5-E15.5 (Figure 4C).
Blood islands (BIs) and coronary vessels in the ventricular septum
BIs were previously reported to arise from endocardium by clonal analysis11. We noticed that the BIs on the ventricle wall were isolated and separated from the surrounding coronary plexus (Figure 6B). We measured the separation distance and found that it was similar to the width of the avascular zone in ventricle wall (Figure 6A–6C). We also observed that BI formation was mostly at the midline near the interface between the septum and ventricle wall (Figure 6E). These structures are connected with the heart lumen (Figure 6D) and clonally related to the endocardium11, raising the possibility that they arise from endocardial budding. BI presence was most prominent on the ventral face of the heart (Figure 6E), which is the last region to be populated by migrating subepicardial vessels (Figures 1A and 4D). In addition, some mutant mice with faulty subepicardial coronary sprouting (Fog2 and VCAM mutants) that do not properly develop coronary vessels exhibit an overproduction and enlargement of ectopic BIs in the ventricle walls (Figure 6F; data not shown). This could indicate a possible compensatory attempt to provide coronary vessels when subepicardial vascular sprouting fails. Such a notion requires additional investigation, but is important to acknowledge during studies of injury and disease.

BI formation and coronary vessels in ventricle septum. (A-C) Penetration distance from endocardium (Endo) and avascular distance from BI. n = 10. (D) Consecutive slides of Sema3dLacZ/+ heart showing BI on epicardium (X-gal+) arises from endocardium and connects with chamber (double red arrow). (E) Majority of Endo-derived BIs appear on ventral side of ventricle septum (black lined area). *P < 0.05; n = 42. (F) Expansion of endocardium-derived ECs of BIs in Fog2 mutants. White dotted line indicates impaired SV-derived coronary plexus; white arrows indicate PECAM+ BIs. (G) In remote area of VS, endocardium sinusoids become narrow strand of PECAM+Lectin− cells (white arrowheads), with loss of connection with chamber circulation. White dotted lines outline ventricle septum. Representative of three embryos. Black bar = 0.5 mm; white bar = 100 μm.
Tracing early coronary sprouts revealed a surprisingly striking inheritance pattern in the ventricular septum. While almost all coronary vessels of the ventricle-free wall arose from early subepicardial ECs, very few, if any, vessels in the septum derived from this source (Figures 4D and 5). We first confirmed that PECAM+ cells in the septum were coronary vessels based on Apln expression. Septal ECs, like intramyocardial ECs, but not trabecular endocardium, directly expressed Apln as early as E12.5, and more at E13.5-E14.5 onward (data not shown). In addition, the AplnCreERT2/+ transgene was able to mark septal ECs when tamoxifen induced recombination at either E12.5 or E13.5 (Figure 5 and data not shown). While these septal ECs were clearly not from early traced subepicardial ECs, the definitive origin remains unknown for these de novo formed vessels. It is likely that these vessels developed through trabecular coalescence, as they become sealed off from luminal blood flow. Embryonic hearts injected with FITC-lectin showed that the central portion of the septum was not perfused, and thus, not continuous with the heart lumen, as early as E12.5 (Figure 6G). Direct evidence of the origin of these septal vessels requires additional specific and inducible lineage tracing of ventricular endocardial cells, which merits further investigation.
Coronary arteries and veins have a common developmental origin
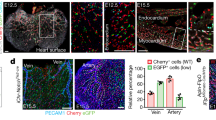
Co-immunofluorescence with arterial- and venous-specific antibodies indicated that E10.5-E11.5 subepicardial ECs produced both intramyocardial coronary arteries and subepicardial veins at E15.5 (Figures 7A and 7B). Consistent with previous work11,12, most coronary arteries were located in the compact myocardium, and coronary veins were located in the subepicardial space (Supplementary information, Figure S9). The ability of a single cell to give rise to both populations has been reported for cells within the SV and endocardium11,12. We went on to test whether subepicardial coronary ECs that had migrated onto the heart retained this ability. Clonal analysis was performed using the AplnCreERT2/+ driver paired with the Confetti multicolor reporter Rosa26Rainbow/+20. The advantage of this reporter allele is that it is very resistant to recombination, and, when recombination occurs, one out of four different permanent labels is selected. Out of 85 AplnCreERT2/+;Rosa26Rainbow/+ hearts, only 14 hearts were labeled with a low dose of tamoxifen, instilling high confidence in their clonality, particularly as most had 1-2 isolated clusters per heart (Figure 7C and 7D). The Confetti reporter Rosa26Rainbow/+ never exhibited tamoxifen-independent activity (data not shown). Each of the clones, which were labeled when only subepicardial cells were present, contained offspring in both the subepicardial and intramyocardial locations, but never endocardium (Figure 7E and 7F). The subepicardial and intramyocardial sister cells expressed venous and arterial markers, respectively (Figure 7F). In addition, the cells were distributed equally among the arterial and venous compartments when cell numbers were normalized to total number of subepicardial and intramyocardial vessels (Figure 7G, total distribution from Figure 1C).

Single subepicardial EC contributes to both coronary arteries and veins. (A, B) E10.5-labeled subepicardial ECs give rise to arterial cells (NOTCH1+, red arrowheads) and venous cells (COUP TFII+, green arrowheads) at E15.5. White arrowheads indicate COUP TFII+ endocardial cells. (C) Schematic representation of clones recovered from AplnCreERT2/+;Rosa26Rainbow/+ embryos with low-efficiency recombination. (D, E) Representative coronary vessel clone in an E15.5 AplnCreERT2/+;Rosa26Rainbow/+ heart shown in whole mount (D) and section (E). (F) Coronary ECs in one clone were positive for both arterial (NOTCH1) and venous (COUP TFII) markers. Yellow arrowheads indicate subepicardial venous EC (RFP+;COUP TFII+); white arrowheads point to intramyocardial arterial ECs (RFP+;NOTCH1+). (G) Percentage of intramyocardial and subepicardial coronary ECs within single AplnCreERT2/+;Rosa26Rainbow/+ clone compared with the distribution of total vessels. *P < 0.05; n = 5 for all vessels; n = 28 for clonal vessels. (H) VE-Cad-CreERT2-induced coronary clones (white) containing sister cells within the AV junction are distributed throughout the thickness of the heart (double yellow arrow). White dotted line indicates epicardium; red dotted line indicates border between trabecular and compact myocardium. (I) Quantification of the percentage of subepicardial (Subepi., blue) and intramyocardial (Intramyo., red) ECs within each clone at E13.5. A similar graph demonstrating the distribution of total vessels in E13.5 left ventricle (10×) is shown for comparison. n = 3-6. LV, left ventricle; RV, right ventricle; VS, ventricular septum. Yellow bar = 100 μm; white bar = 0.5 mm.
We next reanalyzed a previously published data set of VE-Cad-CreER-induced SV-derived coronary clones to determine the distribution of their progeny across the subepicardial or intramyocardial locations11. Similar to the AplnCreERT2/+-induced clones, sister cells were distributed throughout the thickness of the heart (Figure 7H). The average spread was 30% and 50% in the subepicardial and intramyocardial compartments, respectively, with the remaining 20% in the SV (Figure 7I). This is similar to the distribution pattern of total vessels at this stage of development (E13.5) (total distribution from Figure 1C). In both AplnCreERT2/+- and VE-Cad-CreER-induced clones, sister cells were never confined to just the subepicardial or intramyocardial compartment, but were always present in both compartments.
Lineage tracing of other cardiac cell types: few to no contributions to coronary arteries
Several Cre-expressing mouse lines are available, which label various cardiac cell types including neural crest (Wnt1-Cre), (pro)epicardium (Wt1-Cre, Tbx18-Cre), the first and second heart fields (Nkx2-5) and the second heart fields (Mef2c-Cre, Isl1-Cre, Tbx1-Cre), the latter of which also marks a subset of the right ventricle endocardium. Lineage-tracing experiments with these Cre lines excluded a significant contribution from any of these cell types to coronary vessels within the ventricular-free wall (Supplementary information, Figure S10). Given that previous studies have identified the endocardium as a source of coronary vessels11,12, we quantified cells derived from the second heart field that labeled right ventricle endocardium. Three different Cre lines, Mef2c-Cre, Isl1-Cre, and Tbx1-Cre, were coupled with reporter alleles to permanently label endocardial cells and all their progenies21,22,23 (Figure 8A). These Cre lines labeled a subset of the endocardium within the right ventricle (Figure 8B, 8C and 8F); however, very few coronary ECs in the outer layer of ventricle wall were lineage traced at E15.5 or P7 (Figure 8B–8F). Quantification of the Isl1-Cre experiment showed that 50% of the endocardium was labeled, whereas 3% of coronary vessels were positive for the lineage marker (Figure 8B). This subset was able to give rise to coronary endothelium in other locations as Mef2c-Cre labeled a substantial amount of coronary vessels in the ventricular septum at E15.5 (Figure 8F). Therefore, the small number of labeled coronary vessels in the compact myocardium likely reflects a lack of endocardial contribution, rather than an inability of the subset to chase into the coronary compartment.

Second heart field (Isl1, Tbx1, and Mef2c)-traced endocardial cells do not adopt a coronary EC fate. (A) Schematic illustrating the experimental design for testing the contribution of endocardial cells to coronary vessels in the embryonic ventricle wall. Endocardial cells and their progenies are labeled in red. (B) Isl1-Cre labeled NFATc1+ endocardial cells at E10.5 (yellow arrowheads). Dotted white line outlines the epicardium (left panel). Whereas Isl1-Cre labeled endocardial cells at E10.5, intramyocardial coronary ECs were not labeled at E15.5 (white arrowheads, right panel). Inset shows percentage of Isl1-Cre-traced cells (GFP+PECAM+) in both compartments. CA, coronary artery; Endo., endocardium. *P< 0.05; n = 6. (C, D) Neither Tbx1-Cre nor Mef2c-Cre lineage-traced endocardial cells contribute to intramyocardial ECs in the compact myocardium at E15.5 (white arrowheads). (E) Right lateral whole-mount view of Mef2c-Cre;Rosa26RFP/+ heart shows an unlabeled mature right CA (white arrowheads). (F) Mef2c-Cre labeled endocardial cells and coronary ECs in the VS at E15.5, but very few coronary ECs in the ventricle wall (RV). *P < 0.05; n = 4. White bar = 100 μm; red bar = 1 mm. LV, left ventricle; RV, right ventricle; VS, ventricular septum; Epi, epicardium; Endo., endocardium; Comp., compact layer; Trab., trabecular layer.
Discussion
A recent series of reports studying coronary artery development in mice have challenged the textbook model that describes the (pro)epicardium as the sole source of the heart's vasculature9,10,24. These studies have found that SV sprouting11 and endocardial budding11,12 make a substantial contribution to the mammlian coronary vascular endothelium. Although the (pro)epicardium does not appear to be the primary source of the coronary endothelial layer in mice, it is widely accepted that it gives rise to most coronary smooth muscle and is a potent source of epicardial-derived factors that support coronary growth9,10,15,24. To more completely understand coronary development, we must now define, at the cell population level, the relative contributions of the different sources and how the descendants of each are spatially organized within the entire heart. We developed a new tool to address this issue, the AplnCreERT2/+ mouse line, which allowed us to permanently label only subepicardial ECs and all of their progenies. We found that subepicardial ECs present before E12.5 dramatically expanded and migrated in large numbers into the myocardium of the embryonic ventricle free walls (Figure 9). Clonal analysis revealed that single subepicardial EC gave rise to both coronary veins and intramyocardial coronary arteries/capillaries.

Schematic showing subepicardial ECs as a major source for intramyocardial coronary vessels. During embryogenesis, subepicardial ECs (blue) located beneath the epicardium (brown) migrate along the surface of the heart between E11.5-E12.5 (blue arrow). Subepicardial ECs then migrate into the compact myocardium to become intramyocardial coronary arteries and capillaries at E13.0-E15.5 (red arrows).
Although Apln-driven CreERT2 line could lineage-trace the subepicardial ECs and reveal their fate at later stages, the tool alone could not define the origins of these subepicardial ECs. Clues for the possible origin of these subepicardial vessles come from histological observations and clonal analysis where the vessels appear to migrate in from the venous inflow tract of the heart called the SV11,13. Our in vitro culture experiments where genetically labeled (AplnCreERT2/+;Rosa26RFP/+ or Tie2-Cre;Rosa26LacZ/+) SV/atria were paired with unlabeled ventricles suggested that the endocardium of SV/atria, but not the ventricle, gives rise to the primary coronary vessel plexus. Our results of coronary vessels in ventricle wall are consistent with earlier studies on the SV11, but are in contrast with others, showing that the ventricular endocardium is the primary source of intramyocardial coronary vessels12.
Lineage-tracing work using an Nfatc1-Cre line detected a large ventricle endocardial contribution to intramyocardial coronary vessels12. However, we found little histological evidence of ventricular endocardial cells migrating into the myocardium of the ventricle-free wall in any of the stages examined. Subepicardial sprouts were continuously connected with intramyocardial vessels (Figure 1D). This observation suggests that it is less likely that a substantial number of coronary artery progenitors delaminate from the endocardium and populate the compact myocardium. In the previous study12, clonal analysis revealed that single Nfatc1+ cell gives rise to both coronary arteries and veins, but with greater contributions to the intramyocardial (86%) than the subepicardial fraction (14%)12. However, when this Nfatc1-based clonal data are normalized to the distribution of total vessels within each compartment, 84.4% intramyocardial versus 15.6% subepicardial vessels (Figure 1C), the relative distribution of Nfatc1-derived cells of single clone would be almost equal for arteries and veins. Thus, Nfatc1-inducible Cre-mediated clonal analyses seem to reveal that single Nfatc1-positive cell contributed to both arterial and venous ECs with similar frequency.
Wu et al.12 also performed functional analyses that support a ventricular endocardial origin of intramyocardial coronary arteries. Deleting VEGFR2, a receptor required for EC growth, using Nfatc1-Cre resulted in a decrease in coronary vessel development that was more dramatic in intramyocardial vessels. One caveat to these experiments is that thinned myocardium was also present, which is known to specifically affect the intramyocardial vessel fraction25,26. Another issue is that Nfatc1-Cre labels part of the endocardium of SV and atrium in addition to that of the ventricle12, and it was also reported that Nfatc1 itself was expressed in the proximal SV27. As atrial, ventricular, and SV endocardial cells are all labeled by Nfatc1-Cre, we reason that Nfatc1-derived coronary arteries could not be simply interpreted as originating from ventricular endocardium by direct migration into compact myocardium12. It remains equally possible that, instead of ventricular endocardium, these vessels derive from the endocardium of the atrium and SV11.
Difference in conclusions in the origins of coronary arteries may have resulted from the difficulty of finding promoters that are expressed only in the cell population of interest. This can be particularly challenging in the developing heart where the proposed three sources (PEO, SV, and endocardium) for coronary vessels are all adjacent to each other and sometimes continuous. For example, Sema3d-Cre proepicardial cells also lineage-label a proportion of the SV and endocardium7. In addition, the SV neighbors and is the precursor of some atrial endocardial cells, making it not surprising that Nfatc1-Cre traces a subset of the SV12,27. A solid understanding should emerge from multiple studies by independent experiments and more specific tools, such as Cre lines highly specific to an individual component. Inducible Cre lines could also be helpful in circumventing the inherent limitations of their constitutive counterparts8,28,29,30.
In our study, we bypass some of these issues by carrying out lineage-tracing experiments at a stage (E10.5-E11.5) when descendent cell types are more spatially separated. Specifically, we focused on the behavior of early subepicardial ECs by using an inducible AplnCreER. We show that nascent subepicardial ECs massively expand and migrate into the compact myocardium where they form a large number of intramyocardial coronary arteries/capillaries, in addition to coronary veins (Figure 9). Unexpectedly, there were region-specific effects. Subepicardial ECs did not give rise to many of the coronary vessels within the ventricular septum. This new mouse line and experimental regimes described here should be useful in studying coronary vessel behavior during postnatal growth and in the adult during injury, regeneration, and diseases.
Materials and Methods
Generation of AplnCreERT2/+ and Sema3dLacZ/+ knockin lines
A 129-mouse BAC clone containing the complete mouse Apln gene was obtained from Sanger Institute (UK). A targeting vector was constructed using the following fragments: CreERT2 cDNA, SV40 polyA sequence, and PGK-EM7-Neo-resistant gene flanked by Frt sites. Two homologous arms on 5′ and 3′ sides of the first coding exon of Apln gene were generated on targeting vectors by recombination from BAC. The targeting vector was digested with I-CeuI for linearization, and then electroporated in mouse ES cells. The targeting vector was knocked into Apln locus for endogenous expression of CreERT2 cDNA (Supplementary information, Figure S1). After G418 selection, 192 clones were selected for retrieval of genomic DNA and for screening of positive clones. Neomycin-resistant clones were tested for correct gene targeting by long PCR assays, with primer pairs spanning the targeting vector and flanking genomic DNA. Correct ES-targeted clones were confirmed by Southern blot using 5′ and 3′ probes. Following verification of correct targeting and karyotype, positive clone ES cells were expanded and injected into blastocyst for mice generation. The obtained chimera mouse lines were crossed to C57B6 lines for germline transmission. PCR was carried out to test the correct genotype, and the established AplnCreERT2/+ mice were maintained on C57B6 background.
For Sema3dLacz/+ knockin mice, we obtained the targeting vector from KOMP. This targeting vector uses knockout-first strategy, in which Sema3d transcription is blocked before Flp recombination in the targeted mice, and LacZ cDNA is expressed as surrogate for Sema3d mRNA (Supplementary information, Figure S2). After Flp recombination, it could be switched into conditional allele. Targeting vector was linearized by AsiSI and electroporated into mouse ES cells for G418 selection. The following screening step, ES cell injection into blastocyst, and germline transition were same as AplnCreERT2/+ mouse generation. Germline-transmitted mice were crossed to C57B6 lines for maintenance.
Mice
All mice were used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. AplnCreERT2/+ and Sema3dLacZ/+ mice were constructed by knocking exogenous cDNA into Apln or Sema3d locus. Mice were maintained on a C129/C57BL6/J-mixed background. Tamoxifen (Sigma) was dissolved in corn oil and introduced by gavage at the indicated time (0.1-0.15 mg/g). For clonal analysis, dams received an injection of tamoxifen 0.25-0.5 mg dissolved in corn oil. AplnLacZ/+16, Rosa26LacZ/+17, Rosa26mTmG/+18, Rosa26RFP/+19, Rosa26Rainbow/+20, Fog2-mut31, Tie2-GFP32, Tie2-Cre33, Wnt1-Cre34, Nkx2-5Cre/+35, Wt1GFPCre/+10, Wt1CreERT2/+10, Islet1Cre/+9, Mef2c-Cre23, Tbx1-Cre22, VE-Cad-CreERT236, Tbx18-Cre9, EphB4LacZ/+37, and Efnb2LacZ/+38 mice were described previously.
In situ hybridization
Whole-mount in situ hybridization was performed as previously described39. Briefly, whole dissected embryos were fixed overnight in 4% paraformaldehyde. Fixed embryos were dehydrated in a gradient concentrations of methanol and could be stored at −20 °C for a couple of weeks. After dehydration, the embryos should be rehydrated and digested with Proteinase K at a concentration of 20 μg/ml. Embryos were re-fixed with 0.2% glutaraldehyde and 4% paraformaldehyde for 20 min, followed by a wash with 0.1% Tween 20 in PBS and incubation in hybridization buffer for 1 h at 70 °C. Embryos were transferred into fresh hybridization buffer containing 1 μg/ml digoxigenin UTP-labeled RNA probes and incubated overnight with rocking at 70 °C. After washing in a serial of wash solution buffers and treating with ribonucleases, the embryos were incubated with anti-digoxigenin alkaline phosphatase (AP)-conjugated antibody (Roche) overnight with rocking at 4 °C. Next day, embryos were washed with 2 mM levamisole in TBST for a whole day to remove the residual antibody and to inhibit endogenous AP activity. AP activity for probe detection was developed in BM purple (Roche) in dark until the color has been developed to the desired extent. For in situ hybridization on cryosection, dissected embryos were fixed overnight in 4% paraformaldehyde and embedded in optimal cutting temperature (OCT, Sakura). Embryos were sectioned at 6-10 μm thick. Slides were digested with Proteinase K, acetylated with triethanolamine, dehydrated, and hybridized overnight with probes. The slides were then washed in SSC buffer and treated with ribonucleases at 37 °C for 30 min. After incubation with anti-digoxigenin antibody, the slides were developed with BM purple in dark.
Immunohistochemistry
Immunostaining was performed as previously described40. Embryos and adult hearts were collected in PBS on ice and then fixed in 4% PFA at 4 °C for 2 h to overnight. After washing in PBS, tissues were treated sequentially with 15% and 30% sucrose until tissue was fully penetrated. Then they were embedded in OCT (Sakura) and snap frozen. Cyrosections of 6-10 μm thickness were collected on positively charged slides. Tissues were blocked with PBS supplemented with 0.1% Triton X-100 and 5% normal donkey serum (NDS, Jackson ImmunoResearch) for 1 h at room temperature, followed by first antibody incubation at 4 °C for 2 h to overnight. Signals were developed with Alexa fluorescence antibodies (Invitrogen). For weak signals, we used HRP- or biotin-conjugated antibodies with tyramide signal amplification kit (PerkinElmer). Before coverslip addition, tissues were counterstained with 4' 6-diamidino-2-phenylindole (DAPI, Vector lab). Images were acquired by Zeiss confocal microscope LSM510, Leica stereo microscope M165 FC, and Olympus microscope BX53. The sources and dilutions of antibodies used in this study are listed in Supplementary information, Table S1.
Whole-mount PECAM staining
Whole-mount PECAM staining was performed as previously described41. Briefly, embryonic hearts at different time points were collected and fixed in Zinc fixative (Becton Dickinson) overnight at room temperature or 4% fresh PFA overnight at 4 °C. Embryonic hearts were washed in PBS three times, followed by serial dehydration by methanol. Samples were bleached in 5% hydrogen peroxide in 100% methanol for 2 h at 4 °C to inhibit endogenous peroxidase activity, followed by rehydration. Samples were blocked in PBS containing 5% NDS and 0.1% Triton X-100 for 1 h at 4 °C. Samples were then incubated in block solution containing PECAM antibody (BD) overnight at 4 °C, followed by five times wash in PBS. Then, samples were incubated with secondary antibody anti-rat-biotin (BD) in block solution for 12 h, followed by five times PBS wash. Finally, samples were incubated with ABC (Vector Lab) for 2 h at room temperature and washed in PBS for five times, then developed by chromogen AEC or DAB (Vector Lab) at room temperature.
X-gal staining
X-gal staining was performed as previously described41. Hearts were fixed in LacZ fix solution (0.2% glutaraldehyde, 5 mM EGTA, and 100 mM MgCl2 in PBS) for 15-30 min based on the size. After washing three times for 15 min in LacZ wash buffer containing 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40 in 100 mM sodium phosphate buffer, specimens were stained overnight at 37 °C in LacZ stain solution containing 1 mg/ml 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-gal) in LacZ wash buffer. Pictures were taken under Leica stereo microscope M165 FC.
Ex vivo explant assay
Organ culture protocol was followed as described, with minor modifications11. E11.5 embryonic hearts were from Tie2-Cre;Rosa26LacZ/+ or AplnCreERT2/+;Rosa26RFP/+ mice. In brief, mutant SV/A and ventricle were separated by fine forceps and recombined with WT ventricle and SV/A. The recombined hearts were placed dorsal side up at air-liquid interface on Millicell-CM inserts (Millipore). Cultures were conducted at 37% and 5% CO2 for 72 h in DMEM supplemented with 10% fetal bovine serum, 200 U/ml penicillin, 200 μg/ml streptomycin, 0.292 mg/ml L-glutamine, 0.25 μg/ml amphotericin B, 0.25 U/ml heparin, and 100 nM 4-hydorxytamoxifen (Invitrogen). Hearts from Tie2-Cre;Rosa26LacZ/+ mice were cultured in medium without 4-hydroxytamoxifen and subjected to whole mount X-gal staining afterwards.
Statistical analysis
Data were analyzed by unpaired Student's t-tests for two groups, and ANOVA test for over two groups. Significance was accepted when P < 0.05. All data are presented as mean ± SEM.
References
Riley P, Smart N . Vascularizing the heart. Cardiovasc Res 2011; 91:260–268.
Riley P . Developmental biology: Plumbing the heart. Nature 2010; 464:498–499.
Hutchins G, Kessler-Hanna A, Moore G . Development of the coronary arteries in the embryonic human heart. Circulation 1988; 77:1250–1257.
Bogers A, Gittenberger-de Groot A, Poelmann R, Peault B, Huysmans H . Development of the origin of the coronary arteries, a matter of ingrowth or outgrowth? Anat Embryol (Berl) 1989; 180:437–441.
Majesky M . Development of coronary vessels. Curr Top Dev Biol 2004; 62:225–259.
Perez-Pomares J, Phelps A, Sedmerova M, et al. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev Biol 2002; 247:307–326.
Katz Tamar C, Singh Manvendra K, Degenhardt K, et al. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev Cell 2012; 22:639–650.
Merki E, Zamora M, Raya A, et al. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci USA 2005; 102:18455–18460.
Cai C, Martin J, Sun Y, et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008; 454:104–108.
Zhou B, Ma Q, Rajagopal S, et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008; 454:109–113.
Red-Horse K, Ueno H, Weissman I, Krasnow M . Coronary arteries form by developmental reprogramming of venous cells. Nature 2010; 464:549–553.
Wu B, Zhang Z, Lui W, et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF Signaling. Cell 2012; 151:1083–1096.
Bennett HS . The development of the blood supply to the heart in the embryo pig. Am J Anat 1936; 60:27–53.
Poelmann R, Gittenberger-de Groot A, Mentink M, Bokenkamp R, Hogers B . Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ Res 1993; 73:559–568.
Lavine K, Long F, Choi K, Smith C, Ornitz D . Hedgehog signaling to distinct cell types differentially regulates coronary artery and vein development. Development 2008; 135:3161–3171.
Sheikh A, Chun H, Glassford A, et al. In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol Heart Circ Physiol 2008; 294:H88–98.
Soriano P . Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 1999; 21:70–71.
Muzumdar M, Tasic B, Miyamichi K, Li L, Luo L . A global double-fluorescent Cre reporter mouse. Genesis 2007; 45:593–605.
Madisen L, Zwingman T, Sunkin S, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010; 13:133–140.
Snippert H, van der Flier L, Sato T, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010; 143:134–144.
Milgrom-Hoffman M, Harrelson Z, Ferrara N, et al. The heart endocardium is derived from vascular endothelial progenitors. Development 2011; 138:4777–4787.
Huynh T, Chen L, Terrell P, Baldini A . A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis 2007; 45:470–475.
Verzi M, McCulley D, De Val S, Dodou E, Black B . The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol 2005; 287:134–145.
Wilm B, Ipenberg A, Hastie N, Burch J, Bader D . The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 2005; 132:5317–5328.
van den Akker N, Caolo V, Wisse L, et al. Developmental coronary maturation is disturbed by aberrant cardiac vascular endothelial growth factor expression and Notch signalling. Cardiovasc Res 2008; 78:366–375.
Lavine K, Ornitz D . Shared circuitry: developmental signaling cascades regulate both embryonic and adult coronary vasculature. Circ Res 2009; 104:159–169.
Misfeldt A, Boyle S, Tompkins K, et al. Endocardial cells are a distinct endothelial lineage derived from Flk1+ multipotent cardiovascular progenitors. Dev Biol 2009; 333:78–89.
Christoffels V, Grieskamp T, Norden J, et al. Tbx18 and the fate of epicardial progenitors. Nature 2009; 458:E8–E9.
Rudat C, Kispert A . Wt1 and Epicardial Fate Mapping. Circ Res 2012; 111:165–169.
Zhou B, Pu WT . Genetic Cre-loxP assessment of epicardial cell fate using Wt1-driven Cre alleles. Cir Res 2012; 111:e276–e280.
Tevosian SG, Deconinck AE, Tanaka M, et al. FOG-2, a cofactor for GATA transcription factors, is essential for heart morphogenesis and development of coronary vessels from epicardium. Cell 2000; 101:729–739.
Motoike T, Loughna S, Perens E, et al. Universal GFP reporter for the study of vascular development. Genesis 2000; 28:75–81.
Kisanuki Y, Hammer R, Miyazaki J, et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 2001; 230:230–242.
Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP . Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol 1998; 8:1323–1326.
Moses K, DeMayo F, Braun R, Reecy J, Schwartz R . Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 2001; 31:176–180.
Monvoisin A, Alva J, Hofmann J, et al. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn 2006; 235:3413–3422.
Gerety S, Wang H, Chen Z, Anderson D . Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell 1999; 4:403–414.
Wang H, Chen Z, Anderson D . Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998; 93:741–753.
Cai C, Liang X, Shi Y, et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 2003; 5:877–889.
Zhou B, von Gise A, Ma Q, Hu Y, Pu W . Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol 2010; 338:251–261.
Zhou B, Ma Q, Kong S, et al. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J Clin Invest 2009; 119:1462–1476.
Acknowledgements
We thank William Pu, T Quertermous, Sergei Tevosian, M Yanagisawa, Luisa Iruela-Arispe, Hongkui Zeng, David Anderson, and Yosuke Mukoyama for mouse strains, NH Jing for valuable comments, INS facility and animal facility members for technical help. BZ was supported by the Ministry of Science and Technology of China (2012CB945102 and 2013CB945302), Chinese Academy of Sciences (874 and KSCX2-EW-R-09), Shanghai PuJiang Talent program (11PJ1411400), and the National Natural Science Foundation of China (31271552 and 31222038). KR was supported by NIH (4R00HL10579303).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Generation of AplncreERT2/Y knockin mouse line. (PDF 556 kb)
Supplementary information, Figure S2
Generation of Sema3dLacZ/+ knockin mouse line. (PDF 465 kb)
Supplementary information, Figure S3
Characterization of Sema3dLacZ/+ knockin line. (PDF 815 kb)
Supplementary information, Figure S4
X-gal and ESR double staining on E9.5 AplncreERT2/+;Sema3dLacZ/+ embryos show no overlapping signal on proepicardial cells (PEO, white arrowheads) (PDF 622 kb)
Supplementary information, Figure S5
Apln expression map by AplnLacZ/+ line. (PDF 910 kb)
Supplementary information, Figure S6
Fate map of AplncreERT2/+ by Rosa26LacZ/+ reporter. (PDF 693 kb)
Supplementary information, Figure S7
Controls for AplncreERT2/+ fate map. (PDF 841 kb)
Supplementary information, Figure S8
Fate map of AplncreERT2/+ by Rosa26RFP/+ and Rosa26mTmG/+ line. (PDF 1130 kb)
Supplementary information, Figure S9
Beta-GAL and PECAM staining of E 15.0 EphB4LacZ/+ and Efnb2LacZ/+ hearts. (PDF 417 kb)
Supplementary information, Figure S10
Fate mapping of coronary endothelial cells from various cardiac lineages. (PDF 1221 kb)
Supplementary information, Table S1
List of antibodies used in the study. (PDF 120 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article
Cite this article
Tian, X., Hu, T., Zhang, H. et al. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res 23, 1075–1090 (2013). https://doi.org/10.1038/cr.2013.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.83
Keywords
This article is cited by
-
Dual genetic tracing reveals a unique fibroblast subpopulation modulating cardiac fibrosis
Nature Genetics (2023)
-
Hypoxia promotes a perinatal-like progenitor state in the adult murine epicardium
Scientific Reports (2022)
-
Restricted differentiative capacity of Wt1-expressing peritoneal mesothelium in postnatal and adult mice
Scientific Reports (2021)
-
Coordination of endothelial cell positioning and fate specification by the epicardium
Nature Communications (2021)
-
Coronary vessel formation in development and disease: mechanisms and insights for therapy
Nature Reviews Cardiology (2020)
