
Abstract
The INO80 (inositol requiring mutant 80) chromatin remodeling complex plays important roles in transcriptional regulation and DNA replication and repair, and consists of several functional protein subunits, including the critical Ino80 ATPase catalytic subunit. While the function of INO80 has been studied in yeast and mammalian cell lines, we do not know how mIno80 contributes to the maintenance of genome stability to prevent cancer development in mice. Here, we use a conditional knockout approach to explore the cellular and organismal functions of mIno80. Deletion of mIno80 results in profound cellular proliferative defects and activation of p21-dependent cellular senescence. While mIno80 is required for efficient repair of DNA double strand breaks, its depletion did not impact upon the formation of γ-H2AX and 53BP1 DNA damage foci, or the activation of the ATM-CHK2-dependent DNA damage response. mIno80 deletion inhibited the generation of single-strand DNA, resulting in defects in homology-directed DNA repair (HDR) at telomeres. Fragile telomeres were prominent in mIno80Δ/Δ MEFs, suggesting that chromatin remodeling is required for efficient telomere replication. mIno80−/− mouse embryos die early during embryogenesis, while conditional deletion of mIno80 in adult mice results in weight loss and premature death. In a p53−/− tumor-prone background, mIno80 haploinsufficiency favored the development of sarcomas. Our studies suggest that the mIno80 chromatin remodeling complex plays important roles in telomere replication, HDR-mediated repair of dysfunctional telomeres, and maintenance of genome stability.
Similar content being viewed by others


Introduction
DNA double-strand breaks (DSBs) are genotoxic lesions generated exogenously by ionizing radiation or endogenously by stalled DNA replication forks. If these lesions are not properly repaired by homology-directed repair (HDR) or non-homologous end joining (NHEJ) repair pathways, genome instability ensues, which is potentially tumor-promoting. Repair of DSBs requires end resection of DNA breaks by 5′ nucleases to generate single-stranded (ss) DNA for binding by NHEJ or HDR repair complexes and DNA damage checkpoint proteins. However, the presence of nucleosomes represents barriers to DNA repair. Studies of the ATP-dependent INO80 chromatin remodeling complex reveal that it modulates the position of nucleosomes along DNA, using the energy from ATP hydrolysis to provide accessibility to chromatinized DNA, not only for transcription and replication but also for DNA damage repair1,2,3,4. INO80 is a member of the SWI/SNF2 superfamily of chromatin remodeling complexes and is highly conserved from yeast to man5,6. The mammalian INO80 complex is composed of at least 13 protein subunits, including the homologous Ino80 catalytic subunit with ATPase activity and many other orthologous subunits with evolutionarily conserved functions as well as unique subunits6,7,8. Ino80 is the core component of this complex and its ATPase domain is required for interactions with other subunits, including Tip49a/b proteins functionally related to the bacterial RuvB helicase essential for DSB repair7,9.
Genetic studies in yeast have shown that the yINO80 complex plays a role in telomere metabolism10. Telomeres are repetitive G-rich DNA-protein complexes that cap the ends of eukaryotic chromosomes and play essential roles in preventing the activation of DNA damage checkpoints that would otherwise induce cellular senescence and/or apoptosis11. Maintenance of telomere function requires the enzyme telomerase and shelterin, a complex of six proteins required to prevent telomeres from inappropriately activating the DNA damage checkpoint function. Shelterin components TRF1 and TRF2 bind to the duplex regions of telomeric DNA, while POT1 binds to the 3′ ss telomeric G-overhang12,13,14,15. Two POT1 proteins, mPOT1a and mPOT1b, are encoded by the mouse genome. Dysfunctional telomeres arising either from the removal of shelterin components or from critical telomere attrition resulting from diminished telomerase activity are recognized as DSBs. mPOT1a/b and TRF2 repress activation of the DNA damage signaling kinases ATR and ATM, respectively, while mPOT1b represses 5′ nucleolytic processing of the telomeric C-strand, limiting the formation of the 3′ ss telomeric overhang that could otherwise serve as a DNA substrate to initiate HDR13,16,17,18. In the absence of telomerase, deletion of the Ies3p subunit of yINO80 resulted in telomere length elongation, formation of extrachromosomal telomeric circles and increased chromosome fusions, suggesting that yINO80 is involved in the regulation of telomere function10. yINO80 also mediates efficient HDR at telomeres10,19.
Recent studies of mammalian INO80 functions by siRNA-mediated knockdown approaches revealed that this complex plays critical roles in the repair of DSBs and maintenance of genome stability20,21,22,23,24. However, little is known about INO80's function at mammalian telomeres. To investigate the functions of the INO80 complex in the maintenance of telomere homeostasis, we generated a knockout mouse model to conditionally delete mIno80 in cells as well as in the mouse. Deletion of mIno80 results in early embryonic lethality, indicating that mIno80 is essential during mouse embryogenesis. mIno80 is not required to mediate the ATM-CHK2 DNA damage response (DDR), but is critical for the generation of ssDNA to promote HDR of dysfunctional telomeres. In addition, mIno80 deficiency induced telomere replication defects, demonstrating that telomere replication requires chromatin remodeling. Finally, mIno80+/−; p53−/− mice develop a striking shift in tumor spectrum from lymphomas to sarcomas, suggesting that mIno80 plays a role in determining the tumor types generated. Our results shed light on how mIno80 functions to promote telomere replication, repair and maintenance of genome stability.
Results
mIno80 is an essential gene required for early mouse embryonic development
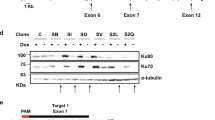
To explore the in vivo functions of mINO80 complex in mice, a Cre-LoxP conditional knockout strategy was employed to disrupt this complex in vivo. We deleted the mIno80 gene by flanking both exons 3 and 4 with loxP sites (Figure 1A and Supplementary information, Figure S1A and S1B). Cre-mediated deletion of these exons results in a frameshift mutation, leading to premature termination of the mIno80 open reading frame. An out-of-frame 52-amino acid peptide that does not bear any sequence similarity to the mIno80 protein could be generated with this approach, but is not expected to possess any functions (Supplementary information, Figure S1A and S1B). Two mouse lines containing the targeted mIno80 floxed allele (mIno80F) were crossed with two Cre-expressing transgenic mouse lines: ZP3-Cre mice to delete the mIno80 gene directly in the germ line to generate mIno80−/− embryos, and CAG-CreER mice to enable conditional deletion of the mIno80F/F alleles (generating mIno80Δ/Δ alleles) in tissues of adult mice by the administration of tamoxifen (TAM)25. We also generated CAG-CreER; mIno80F/F mouse embryo fibroblasts (MEFs) and confirmed conditional deletion of mIno80 upon addition of 4-hydroxytamoxifen (4-HT) to the culture medium (Figure 1B). Western blot and real-time PCR (RT-PCR) analyses both revealed efficient Cre-mediated mIno80 deletion (Figure 1C and 1D). By crossing mIno80+/F mice with the Zp3-Cre mice, we generated mIno80+/− mouse. mIno80+/− intercrosses did not generate any mIno80−/− pups, suggesting that mIno80 deficiency results in embryonic lethality (Figure 1E). Timed pregnancy matings of mIno80+/− mice revealed that all E7.5-E8.5 mIno80−/− embryos exhibited intrauterine developmental abnormalities, including severe growth retardation (Supplementary information, Figure S1C). We found that the majority of mIno80−/− embryos were resorbed by E13.5 to E14.5 (Figure 1E and Supplementary information, Figure S1C). Taken together, our data suggest that mIno80 plays essential roles during mouse embryogenesis.

Targeting strategy for conditional deletion of mIno80. (A) Conditional disruption of the mIno80 ATPase gene. The WT mIno80 allele, targeting construct, targeted mIno80F allele after homologoues recombination, and the null (mIno80Δ) allele following Cre-mediated recombination are shown. The ATG start site is indicated, with exons presented as black boxes, loxP sites as shaded arrowheads, and FRT sites as open arrowheads. (B) Genotyping by PCR analysis. The top panel presents PCR fragment of 375 and 550 bp for WT and mIno80Δ alleles, respectively. The bottom panel presents PCR fragment of 450 and 550 bp for mIno80F and mIno80Δ alleles, respectively. (C) Immunoblotting for mouse mIno80 protein in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment for 5 days. An asterisk indicates a nonspecific band and γ-tubulin levels served as a control for equal protein loading. (D) Real-time PCR analysis for mIno80 mRNA levels in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment for 5 days. Values are normalized to GAPDH expression levels. (E) Genotypes of mice and embryos derived from mIno80+/− intercrosses. The number of the genotypes of postnatal day 21 mice and embryos at E7.5 to 10.5 obtained are indicated.
mIno80 is required for efficient cellular proliferation
To explore the cellular functions of mIno80, early-passage primary CAG-CreER; mIno80F/F MEFs were either untreated, or exposed to 4-HT to delete mIno80. These cells were then subjected to the standard NIH 3T3 proliferation and BrdU incorporation assays. Compared to 4-HT-treated wild-type (WT) or CAG-CreER; mIno80F/F MEFs, the proliferative capacity of CAG-CreER; mIno80Δ/Δ MEFs was significantly impaired. CAG-CreER; mIno80Δ/Δ MEFs stopped proliferating 4 days after 4-HT administration (Figure 2A). Compared to CAG-CreER; mIno80F/F MEFs, BrdU incorporation was also significantly reduced in CAG-CreER; mIno80Δ/Δ MEFs after three passages (45.5% for CAG-CreER; mIno80F/F MEFs vs 4.2% for CAG-CreER; mIno80Δ/Δ MEFs, P < 0.001) (Figure 2B and 2C). These results suggest a defect in both S-phase progression and DNA replication in the absence of mIno80, consistent with previous results observed in Ino80-depleted human cells22. In addition, compared to CAG-CreER; mIno80F/F MEFs, a higher percentage of CAG-CreER; mIno80Δ/Δ MEFs display increased staining at early passages for the senescence marker SA-β-galactosidase (1.8% senescent cells at passage (P) 3 and 1.7% at P4 for CAG-CreER; mIno80F/F MEFs vs 14.7% (P3) and 26.9% (P4) for CAG-CreER; mIno80Δ/Δ MEFs) (Figure 2D). The increased SA-β-galactosidase activity in CAG-CreER; mIno80Δ/Δ MEFs correlated with a robust induction of p21 expression, suggesting premature entry into cellular senescence/cell cycle arrest in the absence of mIno80 function (Figure 2E).

Defects in cellular proliferation and accelerated senescence in mIno80Δ/Δ MEFs. (A) 3T3 proliferation assay of primary MEFs of the indicated genotypes either untreated or treated with 4-HT for 48 h. Three independent cell lines in each genotype were used. (B) Representative images of immunostaining with anti-BrdU antibody for 4 h after BrdU incorporation. (C) Quantification of the percentage of cells stained with anti-BrdU antibody from three independent cell lines per the indicated genotype either untreated or treated with 4-HT. Error bars represent s.e.m. and the Student's t-test was used for statistical analysis. (D) Representative images of primary MEFs stained with X-gal to detect SA-β-galactosidase activity (upper panels) and quantification of the percentage of cells with SA-β-galactosidase activity in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment at passages 3 and 4 (lower panel). Error bars represent s.e.m. and asterisks indicate statistically significant differences (*P < 0.05; **P < 0.005, Student's t-test). (E) Immunoblot analysis for mIno80, p21, PCNA, and γ-tubulin levels in primary MEFs at passage 3. Data from three independent mIno80+/+ (lanes 1-3) and CAG-CreER; mIno80F/F (lanes 4-6) MEFs either untreated or treated with 4-HT are shown.
mIno80 is dispensable for the sensing of DSBs but is required for DNA repair in response to genotoxic stress
Previous results from yeast studies suggest that phosphorylated histone variant H2AX (γ-H2AX) recruited yINO80 to DSBs26,27. To address whether the proliferative defects observed in mIno80Δ/Δ MEFs were due to the activation of a DNA damage checkpoint response, we monitored the formation of γ-H2AX- and 53BP1-positive DNA damage foci in SV40- immortalized CAG-CreER; mIno80F/F MEFs either untreated or treated with 4-HT. We were not able to find any significant changes in the formation of 53BP1 and γ-H2AX DNA damage foci in CAG-CreER; mIno80Δ/Δ MEFs, suggesting that deletion of mIno80 does not by itself induce a robust DDR (Supplementary information, Figure S2A). Next, we asked whether mIno80 is involved in mediating the DDR generated by various genotoxic stresses, including ionizing irradiation (IR) and UV irradiation. Deletion of mIno80 did not significantly impact the accumulation of 53BP1 or γ-H2AX DNA damage foci after 1 h and 8 h exposure to 5 Gy IR (Figure 3A and 3B and Supplementary information, Figure S2B and S2C). Activation of the ATM and ATR kinase pathways was also not abrogated in the absence of mIno80, suggesting that mIno80 is not required to mediate DNA damage signaling (Figure 3C). Interestingly, we found that phosphorylated CHK1 increased in CAG-CreER; mIno80Δ/Δ MEFs, even without exogenous DNA-damaging agents, while increased phosphorylation of the ATM target CHK2 was not detected (Figure 3D). These results suggest that mIno80 is required to repress an ATR-dependent DNA damage checkpoint responses in mouse cells.

Impact of mIno80 deletion on DNA damage signaling and repair. (A) Immunostaining for γ-H2AX- and 53BP1-positive DNA damage foci in CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs treated with 4-HT, 8 or 48 h following 5 Gy IR exposure. (B) Quantification of A for percent of cells containing >10 γ-H2AX or 53BP1 foci. An additional time point at 24 h post IR exposure is included. (C) Immunoblot showing amount of phospho (p)-ATM, total and p-CHK2, γ-H2AX, and γ-tubulin present in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment, harvested at 1, 3, 5, 8, 24 h after exposure to 5 Gy IR. C: unirradiated controls. (D) Immunoblotting for p-CHK1, total CHK2, and γ-tubulin levels in CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment for 96 h. (E, F) Immunostaining of CPD (E) and 6-4PP (F) in CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs treated with 4-HT. For CPD staining, cells were fixed at 0, 6, or 24 h after irradiation with 5 J/m2 of UV (254 nm). For 6-4-PP staining, cells were fixed at 0, 1, and 3 h after 20 J/m2 of UV (254 nm) exposure. Quantifications of DAPI-normalized CPD or 6-4PP signals are shown in the lower panels. Error bars represent s.d. derived from more than six independent data points. Asterisks indicate statistically significant differences (***P < 0.0001, Student's t-test).
48 h after exposure to 5 Gy IR, we found increased γ-H2AX- and 53BP1-positive DNA damage foci in CAG-CreER; mIno80Δ/Δ MEFs (35% in CAG-CreER; mIno80Δ/Δ MEFs vs 21% in CAG-CreER; mIno80+/+ MEFs (Figure 3A and 3B). Compared to control cells, sustained ATM and CHK2 phosphorylation were also observed in CAG-CreER; mIno80Δ/Δ MEFs at later time points following IR exposure (Figure 3C). These results suggest that abrogation of mIno80 function resulted in delayed DSB repair. In agreement with this notion, immunoblot analysis revealed that compared to WT cells exposed to 5 Gy IR, expression of γ-H2AX persisted in the irradiated CAG-CreER; mIno80Δ/Δ MEFs at later time points (Figure 3C). Taken together, our data are in accord with a previous report documenting that mammalian Ino80 is dispensable for the sensing of IR-induced DSBs, but instead is required for IR-induced DSB repair21.
To further explore the role of mIno80 in DNA repair, we analyzed the efficiency of the removal of UV-induced nuclear photo lesions, which are mediated by the nucleotide excision repair (NER) pathway28. Cyclobutane pyrimidine dimers (CPD) and 6-4 photoproducts (6-4PP) are the two primary lesions generated by UV irradiation. Their removal from genomic DNA is a direct indicator of the efficiency of NER repair. We subjected CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs to UV exposure following 4-HT treatment. The amount of photo lesions in each sample was visualized by immunofluorescence staining, using antibodies against CPD and 6-4PP. As shown in Figure 3E, CAG-CreER; mIno80+/+ MEFs exhibited a significant decrease (> 60%) in CPD-positive nuclei 24 h after UV exposure, consistent with the established kinetics of CPD repair. In contrast to these cells, CAG-CreER; mIno80Δ/Δ MEFs retained strong CPD staining after 24 h (Figure 3E, P = 0.0001). Similarly, repair of 6-4PP was also significantly impaired in CAG-CreER; mIno80Δ/Δ MEFs when compared to WT cells (Figure 3F, P = 0.0007 at 1 h and P = 0.0001 at 3 h), suggesting a significant reduction in the efficiency of UV repair in the absence of mIno8029. Consistent with the delayed removal of UV-induced photo lesions, CAG-CreER; mIno80Δ/Δ MEFs displayed decreased cellular survival 48 h after UV exposure (Supplementary information, Figure S2D). Taken together, these results suggest that loss of mIno80 function results in impaired/delayed repair of both IR- and UV-mediated DNA damage.
Defects in ssDNA formation and telomere replication in mIno80-deficient MEFs
Hypersensitivity of yINO80 mutants to hydroxyurea (HU)-induced replication stress suggests that yINO80 is involved in DNA replication. yINO80 localized to origins of replication, and in response to replication stress, the yINO80 complex has been shown to promote the restart of stalled replication forks2,4,30. HDR is required for replication fork restart, and yINO80 has been shown to recruit Rad51 to process recombination intermediates and resolve stalled replication forks31. HDR requires resection of DSBs in a 5′-to-3′ manner to ssDNA for the recruitment of proteins critical for checkpoint activation and repair32, and yINO80 has been shown to promote 5′-to-3′ DNA end resection at DSBs27,33. To determine whether HDR is compromised in the absence of mIno80, we first treated CAG-CreER; mIno80F/F and CAG-CreER; mIno80Δ/Δ MEFs with HU and the DNA polymerase inhibitor aphidicolin (APH). We found significant hypersensitivity of CAG-CreER; mIno80Δ/Δ MEFs to both HU and APH (Supplementary information, Figure S3A and S3B). As formation of ssDNA is a prerequisite for HDR, we next asked whether mIno80 is required for the generation of ssDNA, using a BrdU staining technique that only detects ssDNA under non-denaturing conditions34,35. Compared to CAG-CreER; mIno80F/F MEFs, we found a significant reduction in BrdU-positive foci in CAG-CreER; mIno80Δ/Δ MEFs (50.5% BrdU-positive foci for CAG-CreER; mIno80F/F MEFs vs 37.1% for CAG-CreER; mIno80Δ/Δ MEFs, P = 0.0039) (Figure 4A and 4B). These results suggest that mIno80 is required for the formation of ssDNA.

mIno80 deletion abrogates ssDNA formation and results in telomere replication defects. (A) Detection of ssDNA by immunostaining for BrdU under non-denaturation conditions. 24 h after 10 μM BrdU administration, CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment were exposed to 1 mM HU for 3 h. Anti-BrdU antibody was used to visualize BrdU-positive foci. (B) Quantification of BrdU-positive foci in A. At least 100 cells were counted in three independent cell lines per genotype. Error bars represent s.e.m. and asterisks indicate statistically significant differences (**P < 0.005, Student's t-test) (C) Immunostaining for phospho-RPA32 foci in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment. Cells were treated with 1 mM Hu or 5 μM APH for 3 h prior to analysis for pRPA32 focus formation. (D) Quantification of pRPA32-positive foci in C. At least 100 cells were counted in two independent experiments. Error bars represent s.e.m. and asterisks indicate statistically significant differences (*P < 0.05; **P < 0.005, Student's t-test). (E) PNA-telomere FISH images of CAG-CreER; mIno80F/F MEFs treated with or without 4-HT to monitor the number of fragile telomeres, indicated by the presence of MTS (arrowhead), after vehicle or 0.5 μM APH treatment. (F) Quantification of MTS observed in E. Error bars represent s.e.m. and asterisks indicate statistically significant differences (*P < 0.05, Student's t-test). (G) Immunoblotting for p-CHK1 and γ-tubulin in 4-HT-treated CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs after treatment with either vehicle (C, control) or 5 μM APH for 3 h. Cells were released into normal medium and harvested at 0, 4 and 24 h post treatment. #2 and #3 refer to two independent Ino80-floxed cell lines.
Replication protein A (RPA) is a heterotrimeric protein complex that binds to ssDNA at stalled and collapsed replication forks, and phosphorylation of the RPA32 subunit by ATR modulates checkpoint responses in response to replication stress36,37. Compared to CAG-CreER; mIno80F/F MEFs, we found a significantly reduced level of phospho-RPA32 foci in CAG-CreER; mIno80Δ/Δ MEFs treated with HU or APH (HU: 49.2% p-RPA32 foci for CAG-CreER; mIno80F/F MEFs vs 27.6% for CAG-CreER; mIno80Δ/Δ MEFs; APH: 42.8% for CAG-CreER; mIno80F/F MEFs vs 14.6% for CAG-CreER; mIno80Δ/Δ MEFs) (Figure 4C and 4D). Together, these results suggest that in the absence of mIno80, formation of ssDNA is compromised, resulting in reduced loading of RPA and corresponding reduction in pRPA32 focus formation.
To further validate the requirement of mIno80 for HDR, we examined the status of telomeres in mIno80-deficient MEFs. Telomeres resemble common fragile sites, difficult-to-replicate regions in the genome that, under conditions of replication stress, generate DSBs due to fork stalling38,39,40,41. Common fragile sites are vulnerable to DNA insertions and deletions, promoting genomic instability39,42. Fragile telomeres manifest as telomeric doublets and aberrant strings of telomeric DNA, collectively termed multiple telomere signals (MTS). We found that deletion of mIno80 resulted in a 1.7-fold increase (P = 0.042) in the number of MTS, suggesting that mIno80 is required for proper telomere replication (Figure 4E and 4F). The addition of 0.5 μM APH to CAG-CreER; mIno80Δ/Δ MEFs resulted in a further significant increase in the number of MTS observed, with up to 10% of telomeres displaying aberrant telomeric signals (Figure 4E and 4F, P = 0.0267). A similar result was observed when very low concentration of APH (0.2 μM) was used (data not shown), suggesting hypersensitivity of CAG-CreER; mIno80Δ/Δ MEFs to the presence of APH and formation of fragile telomeres. Consistent with the observation that MTS represent stalled forks containing unreplicated ssDNA38,40, sustained CHK1 phosphorylation was observed in CAG-CreER; mIno80Δ/Δ MEFs following exposure to APH (Figure 4G). Taken together, our data suggest that mIno80 promotes the formation of ssDNA at DNA breaks in stalled replication forks and is required for efficient replication of telomere repeat sequences.
Impaired HDR of dysfunctional telomeres in mIno80Δ/Δ MEFs
The increased number of MTS observed in mIno80Δ/Δ MEFs, coupled with the telomere functions exerted by yINO80, prompted us to examine the role of mIno80 in telomere maintenance. We performed telomere restriction fragment length Southern analysis on CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs either untreated or treated with 4-HT. Deletion of mIno80 did not appreciably alter total telomere length, suggesting that its absence does not adversely impact telomere length maintenance (Supplementary information, Figure S4A). As yINO80 promotes 5′ end resection at DSBs, we also monitored the status of the 3′ ss telomere overhang in CAG-CreER; mIno80Δ/Δ MEFs by hybridizing telomeric oligonucleotides to native telomeric DNA13. Compared to 4-HT-treated CAG-CreER; mIno80+/+ and -untreated CAG-CreER; mIno80F/F controls, the telomeric 3′ ss overhang signal was not significantly altered in CAG-CreER; mIno80Δ/Δ MEFs (Supplementary information, Figure S4A, middle panel). To further monitor the status of telomeres, we performed 2D gel electrophoresis to determine whether aberrant telomeric secondary structures (for example, formation of t-circles) were generated in the absence of mIno80. 2D gel analysis on genomic DNA isolated from CAG-CreER; mIno80F/F and CAG-CreER; mIno80Δ/Δ MEFs did not reveal any significant increase in aberrant secondary telomeric structures in the absence of mIno80, and confirmed that the amount of the telomeric 3′ ss DNA remained largely unaltered in CAG-CreER; mIno80Δ/Δ MEFs (Supplementary information, Figure S4B). The addition of 0.2 μM APH did not significantly alter these results (Supplementary information, Figure S4B). Taken together, these results suggest that acute deletion of mIno80 did not result in significantly increased resection of the 5′ ends of telomeres to generate long 3′ ss G-overhangs.
We next examined the role of mIno80 in the sensing and repair of dysfunctional telomeres. We rendered telomeres dysfunctional in CAG-CreER; mIno80F/F or CAG-CreER; mIno80Δ/Δ MEFs by either removing endogenous mPOT1a and mPOT1b proteins from telomeres using a dominant negative TPP1 mutant (TPP1ΔRD), or by shRNA-mediated depletion of endogenous TRF213,16,43,44. mIno80 deletion did not impact upon telomere dysfunction-induced DNA damage focus (TIF) formation (Figure 5A and 5B and Supplementary information, Figure 4C and 4D), consistent with our finding that mIno80 deletion did not disrupt IR-induced DNA damage focus formation on genomic DNA (Figure 3).

Decreased telomere sister chromatid exchanges at dysfunctional telomeres in the absence of mIno80. (A) Immunostaining for γ-H2AX-positive dysfunctional telomere-induced DNA damage foci (TIFs) following 72 h expression of TPP1ΔRD in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment. Arrowheads point to TIFs. (B) Quantification of A for percentage of cells with > 4 TIFs. At least 100 cells were counted per genotype. Error bars represent s.e.m. n.s.: P-value does not reach significance. (C) Representative metaphase spreads for chromosome orientation FISH in cells expressing either TPP1ΔRD or vector control in CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment. Arrowheads indicate the presence of telomere sister chromatid exchanges (T-SCEs). (D) Quantification of T-SCEs observed in C. Error bars represent s.e.m. and asterisks indicate statistically significant differences (**P < 0.005, Student's t-test). (E) Immunoblot analysis for expression levels of HA-TPP1ΔRD, p-CHK1, total and p-CHK2, and γ-tubulin in CAG-CreER; mIno80+/+ or CAG-CreER; mIno80F/F MEFs expressing TPP1ΔRD, with or without 4-HT treatment.
We next investigated whether mIno80 is required to repair dysfunctional telomeres. shRNA-mediated depletion of TRF2 resulted in uncapped telomeres that are repaired via the classic non-homologous end joining (C-NHEJ)-mediated DNA repair pathway, generating end-to-end chromosomal fusions that require the activation of the ATM-CHK2 pathway44,45. In contrast, removal of mPOT1a/b from telomeres resulted in increased telomere sister chromatid exchanges (T-SCEs) due to elevated HDR at telomeres13,16,46. In addition, the removal of mPOT1a/b also induced chromosome fusions mediated through the Lig4-independent alternative non-homologous end joining (A-NHEJ) DNA repair pathway44. We found that removal of TRF2 in the setting of mIno80 deficiency resulted in robust end-to-end chromosomal fusions and CHK2 activation, similar to those observed in mIno80-competent control MEFs (Supplementary information, Figures S5A-S5C). A-NHEJ-mediated chromosome fusions were also not perturbed in the absence of mIno80 (Supplementary information, Figure S5A and S5B). In contrast, compared to CAG-CreER; mIno80F/F MEFs, a significant reduction in the formation of T-SCEs was observed in CAG-CreER; mIno80Δ/Δ MEFs after removal of mPOT1a/b (Figure 5C and 5D, P = 0.0019). CHK1 activation was also increased in CAG-CreER; mIno80Δ/Δ MEFs upon removal of mPOT1a/b (Figure 5E). These results reinforce our observation that mIno80 is required for HDR of dysfunctional telomeres.
Reduced transformation potential of mIno80-null cells
In addition to its role in the repair of DSBs, yINO80 is required to maintain genome stability by modulating histone H2A.Z distribution in the genome47 and to promote proper chromosome segregation48. We therefore hypothesized that deletion of mIno80 results in genomic instability, a potentially tumor-promoting event. To examine the role of mIno80 in transformed cell lines, we used SV40 large T antigen (SV40LT) alone or in combination with oncogenic H-Ras (H-RasV12) to transform WT, mIno80F/F and mIno80Δ/Δ MEFs. SV40LT-transformed cell lines were then subjected to cell proliferation and colony-forming assays. Compared to adenovirus-Cre-infected WT MEFs, or Ad-empty-infected mIno80F/F MEFs, decreased proliferation was observed only in mIno80Δ/Δ MEFs (Figure 6A). A corresponding reduction in colony formation was observed in mIno80Δ/Δ MEFs transformed with SV40LT alone or together with H-RasV12 (Figure 6B-6E). Finally, we used the soft agar assay to monitor anchorage-independent growth in transformed cells. Compared to SV40LT/H-RasV12-transformed CAG-CreER; mIno80F/F MEFs, a 9.5-15.6-fold reduction in colony size and number was observed in transformed CAG-CreER; mIno80Δ/Δ MEFs (Figure 6F and 6G). Taken together, these results suggest that abrogation of both p53 and Rb functions by SV40LT, alone or in addition to oncogenic H-RasV12 expression, did not rescue the impairment of cellular proliferation following mIno80 deletion.

mIno80 deficiency inhibits cellular proliferation and anchorage-independent growth in oncogene-transformed cells. (A) Growth curve of SV40LT-transformed MEFs of the indicated genotypes with or without Ad-Cre expression. Three independent cell lines per genotype were examined. (B, C) Representative colony forming assays for CAG-CreER; mIno80+/+ or CAG-CreER; mIno80Δ/Δ MEFs. Colonies were stained with crystal violet and scored. (D, E) Quantification of colonies in B, C. Error bars represent s.e.m. and asterisks indicate statistically significant differences (*P < 0.05; ***P < 0.001, Student's t-test). (F) Representative images of colony growth in soft agar (arrows) from SV40LT/H-RasV12-transformed CAG-CreER; mIno80F/F MEFs with or without 4-HT treatment. (G) Quantification of F. Error bars represent s.e.m. and asterisks indicate statistically significant differences (***P < 0.001, Student's t-test).
mIno80 haploinsufficiency results in postnatal growth defects and increased formation of sarcomas in a p53−/− background
Genetic ablation of mIno80 resulted in early embryonic lethality, precluding us from generating mIno80-null mice to address the impact of mIno80 deletion in adult animals (Figure 1E and Supplementary information, Figure S1C). To circumvent this technical limitation, we injected either vehicle or TAM intraperitoneally 3 times into 6-8-week old CAG-CreER; mIno80F/F mice to systemically delete mIno80 in a majority of tissues25. TAM treatment reduced mIno80 expression in the analyzed tissues, from ∼88% (muscle) to ∼60% (brain) (Supplementary information, Figure 6A). Compared to vehicle-treated CAG-CreER; mIno80F/F controls, conditional depletion of mIno80 in vivo resulted in significantly reduced mouse body weight and lifespan, with 50% of TAM-treated CAG-CreER; mIno80F/F mice succumbing 170 days post treatment (Figure 7A and 7B). The causes of death remain unclear at this time, but an increase in organ pathology or an increased cancer incidence was not observed (data not shown). To explore how mIno80 haploinsufficiency impacted mouse survival, we examined the survival profile of mIno80+/− mice. While mIno80+/− mice were viable to adulthood and appeared grossly indistinguishable from their WT littermates at the time of weaning, ∼21% displayed characteristic defects in the hind limb extension reflex, a phenotype associated with motor neuron deficits in the central nervous system (CNS), at as early as 8 weeks of age (Supplementary information, Figure S6B and S6C)49,50. Compared to mIno80+/+ mice, this phenotype was observed ∼5.3 times more frequently in mIno80+/− mice, and it also correlated with a smaller body size (Supplementary information, Figure S6D). These results suggest that mIno80 plays a role in motor neuron function.

mIno80 haploinsufficiency is associated with postnatal growth defects and altered tumor spectrum in a p53-null background. (A) Body weights of mIno80F/F and CAG-CreER; mIno80F/F mice after tamoxifen (TAM) injection. Error bars represent s.e.m. and asterisks indicate statistically significant weight differences (*P < 0.05; **P < 0.005; ***P < 0.001, Student's t-test). (B) Kaplan-Meier survival analysis of mIno80F/F and CAG-CreER; mIno80F/F mice after TAM injection. The log rank test was used to compare difference in survival (P = 0.0048). (C) Quantification of tumor number and spectrum (thymic lymphomas, B-cell lymphomas in the spleen and lymph nodes as well as all other tumor types) in mIno80+/−; p53−/− mice compared to p53−/− mice. (D) Representative H&E sections of lymphomas and sarcomas in mice of the indicated genotypes. Inset: arrowheads point to pleomorphic giant nuclei. (E) Kaplan-Meier analysis for tumor-free survival of mIno80+/−; p53−/− mice compared to p53−/− mice. The log rank test (P = 0.9422) showed no significant differences in tumor-free survival between the two cohorts.
The function of chromatin remodeling complexes in the pathogenesis of several human cancers has been documented51. For example, mutations in the components of chromatin remodeling complexes including Snf5, Brg1, and Baf57 have been associated with diverse cancers52,53,54. Given our observation that deletion of mIno80 significantly reduced the proliferation of transformed cell lines (Figure 6), we asked whether mIno80 deletion reduced the onset of tumorigenesis in vivo. Compared to vehicle-treated controls, tumor incidence was not significantly altered in TAM-treated CAG-CreER; mIno80F/F mice (data not shown). We next investigated whether mIno80 heterozygosity in a tumor-prone p53-null background impacted tumor formation. As deletion of p53 has been shown to rescue the embryonic developmental defects on the setting of impaired DNA damage signaling and repair55, we first monitored the impact of p53 deletion in the generation of mIno80−/− embryos. We found that deletion of p53 did not delay the early embryonic lethality phenotype observed in mIno80−/− embryos (Supplementary information, Figure S6E and data not shown). We next generated mIno80+/−; p53−/− mice and compared their tumor incidence and spectrum to p53−/− controls. Consistent with previously published reports, we found that 6/10 p53−/− mice developed thymic lymphomas and 3/10 developed lymphomas involving the spleen and lymph nodes56 (Figure 7D). Interestingly, this tumor spectrum was altered in mIno80+/−; p53−/− mice. While 7 out of 20 mIno80+/−; p53−/− mice developed thymic lymphomas, 10 out of 20 mice developed poorly differentiated and highly invasive soft tissue sarcomas (Figure 7C and 7D). Compared to p53−/− mice, both the tumor incidence and latency were not appreciably altered in the mIno80+/−; p53−/− background (Figure 7E). These results suggest that mIno80 haploinsufficiency impacted cancer development by altering a p53−/− tumor spectrum dominated by lymphomas to one favoring the formation of invasive sarcomas.
Discussion
The mINO80 chromatin remodeling complex plays important roles in the regulation of transcription, DNA replication and repair. However, our understanding of how chromatin remodelers contribute to genomic instability and cancer pathogenesis in mammals remains limited. In this study, we generated a mIno80 conditional knockout mouse to explore the impact of mIno80 deletion both in cell lines and in the mouse. Unlike yINO80, we found that mIno80 is not required for the sensing of DSBs. However, mIno80 is required for efficient repair of IR-induced DSBs, UV-induced photo lesions and dysfunctional telomeres. Elevated telomere replication defects, manifested as increased fragile telomeres, were observed in mIno80-null MEFs. Deletion of mIno80 also results in reduced DNA synthesis and activation of a p21-dependent cellular senescence program, suggesting that mIno80 is required for S-phase progression. mIno80 also plays an essential role during mouse embryogenesis, as mIno80-null embryos experience early lethality that cannot be rescued by p53 deletion. mIno80+/−; p53−/− mice develop a striking shift in tumor spectrum from lymphomas to sarcomas, suggesting that mIno80 plays a role in determining the types of cancer that develop in mice. Collectively, our results link telomere repair, genome stability, and tumor suppression to the maintenance of chromatin architectural integrity by mIno80.
Following DNA damage, the yINO80 complex is recruited to DSBs through the direct interaction of its Nhp10 or Arp4 subunit with γ-H2AX26,27,57. yINO80 has also been shown to induce γ-H2AX following exposure to DNA-damaging agents and to modulate downstream DNA damage checkpoint functions26,58. We found that both γ-H2AX and 53BP1 efficiently localized to IR-induced DSBs and to dysfunctional telomeres in mIno80Δ/Δ MEFs, suggesting that mIno80 is not required for the accumulation of γ-H2AX and 53BP1 at DNA damage sites. As the mammalian mINO80 complex lacks Nhp10, this species-specific difference likely renders it dispensable for interacting with γ-H2AX at DSBs21,24,59. Rather, mammalian INO80 appears to require its Arp-8 subunit for recruitment to laser-induced DSBs59. IR- and UV-induced DNA damages persist in mIno80Δ/Δ MEFs, suggesting that mIno80 is required for their efficient repair (Figure 3). In addition, we found that mIno80 is required for the generation of ssDNA to mediate HDR at sites of DSBs (Figure 4), in agreement with previous results showing that siRNA-mediated depletion of hIno80 impairs HDR20,23.
Telomeres represent difficult-to-replicate regions of the genome, and replication forks can stall within telomeric repeats, generating fragile telomeres and a corresponding ATR-dependent DDR40. We found that mIno80 repressed the formation of fragile telomeres, suggesting that regulation of chromatin structure is required for telomere replication. In addition, mIno80 was also required to repress the activation of the ATR-CHK1 pathway. While little is known about how modification of chromatin architecture influences telomere replication, yINO80 has been shown to accumulate at origins of replication to promote the recovery of stalled forks2,4,30,31. yINO80 recruits Rad51 to stalled replication forks to mediate the formation of hemicatenated X-shaped DNA structures, which are resolved by the helicase Sgs131. We speculate that mIno80 plays a similar role at telomeres, and is required to resolve aberrant recombination intermediates during telomere replication. In support of this notion, deletion of yINO80 in telomerase-null cells significantly reduced the emergence of Type 1 survivors, suggesting that yINO80 is required for telomere recombination in a Rad51-dependent manner10,19.
At mammalian telomeres, aberrant HDR between sister telomeres (telomere sister chromatid exchanges) are repressed by POT1, likely due to loading of POT1 to ss telomeric DNA to prevent binding of factors involved in HDR13,16,60. yINO80 promotes 5′ end resection and formation of 3′ ss DNA substrates at DSBs27,33,61. However, aberrant 3′ ss telomeric overhang was not observed in mIno80Δ/Δ MEFs, suggesting that mIno80 does not mediate 5′ end resection at telomeres possessing an intact shelterin complex. We are currently generating mIno80−/−; mPOT1b−/− MEFs to determine whether mIno80 is involved in the resection of the telomeric C-strand following mPOT1b deletion62. Other than aberrant fragile telomeres, we also did not observe an increase in structural chromosomal aberrations in mIno80-null MEFs, in marked contrast to a report documenting chromosomal translocations in human cancer cells when mIno80 is knocked down22. While it is not clear what accounts for this discrepancy in chromosome phenotypes, species-specific differences in mIno80 function, the differential rates of cellular proliferation between primary vs tumor cells and cooperative effects of genetic aberrations in tumor cells with mIno80 depletion likely account for this phenotypic difference.
Rhabdoid tumors (RT) containing biallelic inactivating mutations in the gene encoding SMARCB1 was the first link suggesting that ATPase chromatin remodeling complexes play a role in tumor suppression52. Nearly 35% of Smarcb1+/− mice develop RT by 11 months of age, while conditional inactivation of both Smarcb1 alleles results in 100% of mice developing tumors by 11 weeks. Mutations in the subunits of other chromatin remodeling complexes, including Snf5, Brg1, and Baf57, have also been found in a variety of human cancers52,53,54, suggesting that chromatin remodelers function as tumor suppressors. As mIno80 promotes DNA repair and represses genome instability, we postulate that the mIno80 complex also plays a role in tumor prevention. This notion is supported by the observation that hIno80 and its associated protein YY-1 are required to maintain genome stability by promoting proper chromosome segregation20,22. We found that deletion of mIno80 severely compromised the tumorigenic potential of transformed mouse cell lines by reducing cellular proliferative capacity and the ability to form soft agar colonies in culture (Figure 6). While conditional deletion of mIno80 in mice results in progressive weight loss and early death, we did not observe a significant impact on tumor formation in either conditionally deleted mIno80Δ/Δ or mIno80+/− mice. In a tumor-prone p53−/− background, mIno80 haploinsufficiency did not reduce tumor incidence. However, one striking finding in our study was the observation that mIno80 haploinsufficiency has an impact on the types of tumors generated. We found a shift in tumor spectrum from the B- and T-cell lymphomas characteristic of p53−/− tumors to the invasive sarcomas prevalent in mIno80+/−; p53−/− mice. This shift in tumor spectrum could be due to increased genomic instability stemming from telomere replication defects, an inability to engage in HDR-mediated repair, or alterations in gene transcription involved in tumorigenesis. Common fragile sites are vulnerable to DNA insertions and deletions, promoting an unstable genome that contributes to the formation of some human cancers39,42. We speculate that defects in telomere replication in the absence of mIno80 might lead to similar increases in genomic instability to promote tumorigenesis. Interestingly, a recent report revealed that nucleosome depletion by the mIno80 complex is required for embryonic stem cell differentiation, suggesting that mIno80 plays an important role in the determination of cell fate during development63. It is therefore tempting to speculate that reduction of mIno80 levels in the absence of p53 played a role in modulating the cell fate of cancer-initiating cells, to promote the formation of sarcomas over lymphomas.
Materials and Methods
Antibodies and chemicals
HU, APH, and 4-HT were purchased from Sigma-Aldrich. Antibodies used in this study were anti-pCHK1-S345 (133D3, Cell Signaling Technology), γ-Tubulin (079K4861, Sigma-Aldrich), CHK2 (611570, BD transduction laboratory), pRPA32-S4/S8 (A300-245A, Bethyl Laboratories), p21 (SC-6246, Santa Cruz), PCNA (SC-7907, Santa Cruz), γ-H2AX-S139 (05-636, Millipore), 53BP1 (SC-22760, Santa Cruz), and HA (118K4802, Sigma-Aldrich).
Generation of a conditional mIno80 knockout mouse
Exon 2 and 3 of mIno80 gene were constructed into the pKOII targeting vector, containing the diphtheria toxin gene (DT), the PGK-neo gene flanked by two FRT sequences, and loxP sequences. The proximal 4.1 kb and distal 3.2 kb sequences of the mIno80 gene were then ligated into targeting vector to use for homologous recombination (see Figure 1A). The linearized mIno80 targeting vector was electroporated into ES cells and correctly targeted clones were injected into C57BL/6 blastocysts to obtain chimeric mice. Germ line transmission (mIno80F/+ mice) was confirmed by genomic PCR analysis. mIno80F/+ mice were crossed with transgenic mouse bearing ZP3-Cre transgene (Jackson Laboratory, C57BL/6-Tg(Zp3-cre)93Knw/J) and the mIno80F/+ -ZP3 Cre females were crossed with mIno80F/F males to generate mIno80+/− mouse. Transgenic mice bearing the CAG actin-Cre transgene (Jackson Laboratory, B6-Cg-Tg(CAG-cre/Esr1*)5Amc/J) were used to generate CAG-CreER; mIno80F/F mouse lines. Intraperitoneal tamoxifen injection into mice was performed as previously reported25. All mice were maintained and experiments were conducted in accordance with Yale University IACUC-approved guidelines.
MEFs culture and proliferation assays
Primary MEFs were isolated from E13.5 embryos and maintained in DMEM with 10% FBS according to standard protocols. SV40-transformed MEFs were generated by expressing SV40 large T antigen (SV40LT) in early passage of primary MEFs. The mIno80 gene in CAG-CreER; mIno80F/F MEFs was deleted by adding 100 nM of 4-HT for either 48 or 96 h and cells for the indicated experiments were maintained in 4-HT throughout the duration of the experiment. mIno80 deletion was confirmed by RT-PCR and immunoblotting with anti-Ino80 antibody. The 3T3 proliferation assay was performed on primary MEFs according to standard protocols. Cell proliferation assay using BrdU incorporation was performed by incubating primary MEFs with BrdU (10 μM) for 4 h and then immunostained with anti-BrdU antibody after DNA denaturation with 2N Hcl. Colony forming assay in SV40-transformed MEFs was performed following standard protocols. Briefly, 1 × 103 cells were seeded in 6-well tissue culture plates and either untreated or treated with HU or APH and cultured for 1 week. Colonies were fixed with ice-cold methanol, stained with crystal violet (0.5% in 25% methanol) and counted. To evaluate anchorage-independent growth, the soft agar assay was performed in SV40LT/H-RasV12MEFs. Cells were mixed with agar (0.35%) and plated in 6-well tissue culture dishes containing bottom agar (0.7% in culture media). Cells were replenished with growth media every 2 days and monitored for anchorage-independent growth over 2 weeks. Analysis for UV-induced photo lesions was performed as described previously29.
Monitoring replication stress and telomere replication defects
To test for sensitivity to replication stress, cells were exposed to 0.2 μM APH or 0.05 mM HU for 24 h, replenished with fresh media, and cultured for 1week for cell colony-forming assay64. Cells were treated with 5 μM APH or 1 mM HU for 3 h and immediately fixed for pRPA immunostaining or for immunoblotting65. To measure degree of telomere fragility, cells were treated with APH (either 0.2 or 0.5 μM) for 24 h, colcemide for 4 h, and metaphases were prepared for PNA FISH40.
Telomere assays (TRF Southern blot, 2D gel electrophoresis, PNA FISH, chromosome orientation FISH)
Dysfunctional telomeres were induced by overexpression of either mouse TPP1ΔRD (pQCXIP-HA TPP1ΔRD) or shTRF2 (pSuper-shTRF2) using retroviral infection into MEFs as described previously66. Metaphase chromosome spreads were prepared and telomere PNA-FISH and chromosome orientation-FISH using telomere probes (Cy3-OO-(CCCTAA)3, F1002, and 5′-FAM-OO-(TTAGGG)3-3′, F1005, Panagene) were performed as described previously66. For TRF Southern blot, cells were trypsinized and embedded in 1% agarose gel plugs at a concentration of 1 × 106 cells per plug and TRF Southern blot analysis was performed as described13,41. 2D gel electrophoresis was performed as previously described41.
Immunofluorescence analysis
Telomere-induced foci (TIF) analysis in MEFs was performed by immunostaining for γ-H2AX and 53BP1 as previously described66. To measure pRPA32 foci, cells were pretreated with hypotonic solution (solution I: 10 mM HEPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EDTA, and 0.5% Triton X-100; solution II: 10 mM Tris pH7.4, 10 mM NaCl, 1% Tween 20, and 0.25% sodium deoxycholoate) before fixing cells with 4% PFA. Fixed cells were permeabilized with 1% Triton X-100/0.5% NP-40 for 20 min on ice and blocked with PBG solution (0.5% BSA and 0.2% fish gelatin in PBS), incubated overnight with the primary antibody and for 1 h with the secondary antibody at RT, and mounted with Vectastain with DAPI . For the detection of ssDNA, cells were pre-incubated with BrdU (10 μM) for 24 h prior to1 mM HU treatment, as previously described34,35. BrdU-incorporated ssDNA was detected by immunostaing using anti-BrdU antibody without DNA denaturation, as previously described34,35. At least 20 different images were taken and >100 cells per cell line were analyzed. All images involved in immunofluorescence were captured and processed using a Nikon Eclipse 80i microscope and NIS-element BR 3.10 software.
References
Watanabe S, Peterson CL . The INO80 family of chromatin-remodeling enzymes: regulators of histone variant dynamics. Cold Spring Harb Symp Quant Biol 2010; 75:35–42.
Shimada K, Oma Y, Schleker T, et al. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr Biol 2008; 18:566–575.
Trujillo KM, Osley MA . INO80 meets a fork in the road. Nat Struct Mol Biol 2008; 15:332–334.
Vincent JA, Kwong TJ, Tsukiyama T . ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat Struct Mol Biol 2008; 15:477–484.
Morrison AJ, Shen X . Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol 2009; 10:373–384.
Seeber A, Hauer M, Gasser SM . Nucleosome remodelers in double-strand break repair. Curr Opin Genet Dev 2013; 23:174–184.
Jin J, Cai Y, Yao T, et al. A mammalian chromatin remodeling complex with similarities to the yeast INO80 complex. J Biol Chem 2005; 280:41207–41212.
Conaway RC, Conaway JW . The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci 2009; 34:71–77.
Papin C, Humbert O, Kalashnikova A, et al. 3′- to 5′ DNA unwinding by TIP49b proteins. FEBS J 2010; 277:2705–2714.
Yu EY, Steinberg-Neifach O, Dandjinou AT, et al. Regulation of telomere structure and functions by subunits of the INO80 chromatin remodeling complex. Mol Cell Biol 2007; 27:5639–5649.
O'Sullivan RJ, Karlseder J . Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010; 11:171–181.
Baumann P, Cech TR . Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 2001; 292:1171–1175.
Wu L, Multani AS, He H, et al. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 2006; 126:49–62.
He H, Multani AS, Cosme-Blanco W, et al. POT1b protects telomeres from end-to-end chromosomal fusions and aberrant homologous recombination. EMBO J 2006; 25:5180–5190.
Chan SS, Chang S . Defending the end zone: studying the players involved in protecting chromosome ends. FEBS Letters 2010; 584:3773–3778.
Guo X, Deng Y, Lin Y, et al. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J 2007; 26:4709–4719.
Deng Y, Guo X, Ferguson DO, Chang S . Multiple roles for MRE11 at uncapped telomeres. Nature 2009; 460:914–918.
Denchi EL, de Lange T . Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007; 448:1068–1071.
Hu Y, Tang H-B, Liu N-N, et al. Telomerase-null survivor screening identifies novel telomere recombination regulators. PLoS Genet 2013; 9:e 1003208.
Wu S, Shi Y, Mulligan P, et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol 2007; 14:1165–1172.
Park EJ, Hur SK, Kwon J . Human INO80 chromatin-remodelling complex contributes to DNA double-strand break repair via the expression of Rad54B and XRCC3 genes. Biochem J 2011; 431:179–187.
Hur SK, Park EJ, Han JE, et al. Roles of human INO80 chromatin remodeling enzyme in DNA replication and chromosome segregation suppress genome instability. Cell Mol Life Sci 2010; 67:2283–2296.
Gospodinov A, Vaissiere T, Krastev DB, Legube G, Anachkova B, Herceg Z . Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol Cell Biol 2011; 31:4735–4745.
Kitayama K, Kamo M, Oma Y, et al. The human actin-related protein hArp5: Nucleo-cytoplasmic shuttling and involvement in DNA repair. Exp Cell Res 2009; 315:206–217.
Hayashi S, McMahon AP . Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 2002; 244:305–318.
Morrison AJ, Highland J, Krogan NJ, et al. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004; 119:767–775.
van Attikum H, Fritsch O, Hohn B, Gasser SM . Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004; 119:777–788.
Sancar A . DNA excision repair. Ann Rev Biochem 1996; 65:43–81.
Jiang Y, Wang X, Bao S, et al. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc Natl Acad Sci USA 2010; 107:17274–17279.
Papamichos-Chronakis M, Peterson CL . The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat Struct Mol Biol 2008; 15:338–345.
Falbo KB, Alabert C, Katou Y, et al. Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat Struct Mol Biol 2009; 16:1167–1172.
Mimitou EP, Symington LS . Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 2009; 34:264–272.
Chen X, Cui D, Papusha A, et al. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 2012; 489:576–580.
Raderschall E, Golub EI, Haaf T . Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci USA 1999; 96:1921–1926.
Sartori AA, Lukas C, Coates J, et al. Human CtIP promotes DNA end resection. Nature 2007; 450:509–514.
Shao R-G, Cao C-X, Zhang H, Kohn KW, Wold MS, Pommier Y . Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J 1999; 18:1397–1406.
Liu S, Opiyo SO, Manthey K, et al. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res 2012; 40:10780–10794.
Casper AM, Nghiem P, Arlt MF, Glover TW . ATR regulates fragile site stability. Cell 2002; 111:779–789.
Arlt MF, Xu B, Durkin SG, Casper AM, Kastan MB, Glover TW . BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol Cell Biol 2004; 24:6701–6709.
Sfeir A, Kosiyatrakul ST, Hockemeyer D, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009; 138:90–103.
Gu P, Min JN, Wang Y, et al. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J 2012; 16:2309–2321.
Yunis JJ, Soreng AL . Constitutive fragile sites and cancer. Science 1984; 226:1199–1204.
Liu D, O'Connor MS, Qin J, Songyang Z . Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem 2004; 279:51338–51342.
Rai R, Zheng H, He H, et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J 2010; 29:2598–2610.
Dimitrova N, Chen Y-CM, Spector DL, de Lange T . 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008; 456:524–528.
Bailey SM, Brenneman MA, Goodwin EH . Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res 2004; 32:3743–3751.
Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL . Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 2011; 144:200–213.
Chambers AL, Ormerod G, Durley SC, et al. The INO80 chromatin remodeling complex prevents polyploidy and maintains normal chromatin structure at centromeres. Genes Dev 2012; 26:2590–2603.
Barneoud P, Lolivier J, Sanger DJ, Scatton B, Moser P . Quantitative motor assessment in FALS mice: a longitudinal study. Neuroreport 1997; 8:2861–2865.
Weydt P, Hong SY, Kliot M, Moller T . Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport 2003; 14:1051–1054.
Wang GG, Allis CD, Chi P . Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med 2007; 13:373–380.
Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998; 394:203–206.
Klochendler-Yeivin A, Picarsky E, Yaniv M . Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol Cell Biol 2006; 26:2661–2674.
Isakoff MS, Sansam CG, Tamayo P, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci USA 2005; 102:17745–17750.
Gao Y, Ferguson DO, Xie W, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000; 404:897–900.
Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4:1–7.
Downs JA, Allard Sp, Jobin-Robitaille O, et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 2004; 16:979–990.
Papamichos-Chronakis M, Krebs JE, Peterson CL . Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev 2006; 20:2437–2449.
Kashiwaba S, Kitahashi K, Watanabe T, Onoda F, Ohtsu M, Murakami Y . The mammalian INO80 complex is recruited to DNA damage sites in an ARP8 dependent manner. Biochem Biophys Res Commun 2010; 402:619–625.
Flynn RL, Centore RC, O/'Sullivan RJ, et al. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011; 471:532–536.
van Attikum H, Fritsch O, Gasser SM . Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 2007; 26:4113–4125.
He H, Wang Y, Guo X, et al. Pot1b deletion and telomerase haploinsufficiency in mice initiate an ATR-dependent DNA damage response and elicit phenotypes resembling dyskeratosis congenita. Mol Cell Biol 2009; 29:229–240.
Li Z, Gadue P, Chen K, et al. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 2012; 151:1608–1616.
Buonomo SBC, Wu Y, Ferguson D, de Lange T . Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol 2009; 187:385–398.
Groth A, Corpet A, Cook AJ, et al. Regulation of replication fork progression through histone supply and demand. Science 2007; 318:1928–1931.
Rai R, Li JM, Zheng H, et al. The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere end protection. Nat Struct Mol Biol 2011; 18:1400–1407.
Acknowledgements
This work was supported by funding from NCI (RO1 CA129037 to SC; CA127945 and CA097175 Project 3 to LL).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Deletion of mouse mIno80 gene. (PDF 452 kb)
Supplementary information, Figure S2
DNA damage foci are present in mIno80 deleted cells. (PDF 704 kb)
Supplementary information, Figure S3
(A) Representative colony forming assays of 4-HT treated CAG-CreER; mIno80+/+ and CAG-CreER; mIno80F/F MEFs following exposure to 0.2μM aphidicolin (APH) or 0.05mM hydroxyurea for 24hrs. (PDF 3242 kb)
Supplementary information, Figure S4
mIno80 deletion did not alter total telomere length, G-overhang status, and recruitment 53BP1 to dysfunctional telomeres. (PDF 1349 kb)
Supplementary information, Figure S5
mIno80 deletion does not impact upon C-NHEJ or A-NHEJ mediated DNA repair. (PDF 1548 kb)
Supplementary information, Figure S6
Phenotype of mIno80 heterozygote mice. (PDF 3742 kb)
Rights and permissions
About this article
Cite this article
Min, JN., Tian, Y., Xiao, Y. et al. The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Res 23, 1396–1413 (2013). https://doi.org/10.1038/cr.2013.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.113
Keywords
This article is cited by
-
The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective
Cellular and Molecular Life Sciences (2021)
-
INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer
Oncogene (2017)
-
Epigenetic regulation in cell senescence
Journal of Molecular Medicine (2017)
-
Ino80 is essential for proximal-distal axis asymmetry in part by regulating Bmp4 expression
BMC Biology (2016)
-
Epigenomic regulation of oncogenesis by chromatin remodeling
Oncogene (2016)
