
Abstract
Toll-like receptor 9 (TLR9) senses microbial DNA in the endosomes of plasmacytoid dendritic cells (pDCs) and triggers MyD88-dependent type I interferon (IFN) responses. To better understand TLR9 biology in pDCs, we established a yeast two-hybrid library for the identification of TLR9-interacting proteins. Here, we report that an IFN-inducible protein, phospholipid scramblase 1 (PLSCR1), interacts with TLR9 in pDCs. Knockdown of PLSCR1 expression by siRNA in human pDC cell line led to a 60-70% reduction of IFN-α responses following CpG-ODN (oligodeoxynucleotide) stimulation. Primary pDCs from PLSCR1-deficient mice produced lower amount of type 1 IFN than pDCs from the wild-type mice in response to CpG-ODN, herpes simplex virus and influenza A virus. Following CpG-A stimulation, there were much lower amounts of TLR9 in the early endosomes together with CpG-A in pDCs from PLSCR1-deficient mice. Our study demonstrates that PLSCR1 is a TLR9-interacting protein that plays an important role in pDC's type 1 IFN responses by regulating TLR9 trafficking to the endosomal compartment.
Similar content being viewed by others


Introduction
Plasmacytoid dendritic cells (pDCs) are the professional interferon (IFN)-producing cells of the immune system specialized in recognizing viral RNA and DNA through the endosomal TLR7 and Toll-like receptor 9 (TLR9), respectively1,2,3. TLR9 resides in the endoplasmic reticulum (ER) in association with UNC93B and gp964,5,6. TLR9 travels through the Golgi complex to access CpG DNA in endolysosomes7,8. Recently, another protein, AP-3, was shown to be involved in the ER to endolysosome transition of TLR99. Following cellular exposure to nucleic acids, TLR9 relocates from the ER to the endosomes to engage with their DNA agonists10. In order for TLR9 to signal, the ectodomain of TLR9 must be cleaved first in the endolysosomes by cathepsins7,11. TLR9 signals through the adapter protein MyD8812,13, leading to two different pathways. The first pathway leads to the activation of type I IFN genes through phosphorylation of IFN regulatory factor 7 (IRF7)14. The second pathway leads to the transcriptional activation of proinflammatory cytokines through activation of nuclear factor κB (NF-κB)15. Retention of multimeric A-type CpG DNA (CpG-A) in early endosome (EE) is associated with IFN production in pDCs, whereas rapid translocation of monomeric B-type CpG DNA (CpG-B) to the lysosome is associated with NF-κB activation16,17,18. These studies suggested that the intracellular location of TLR9 activation could dictate the nature of its downstream signaling. Although these studies identify a number of molecules involved in TLR9 signaling, the mechanism by which TLR9 triggers IRF7 activation versus NF-kB activation in pDCs remains poorly defined.
Because direct biochemical characterization of TLR9-interacting proteins has been hampered by the rarity of pDCs in human peripheral blood (< 1%), we made a cDNA expression library in the yeast two-hybrid system from 100 million primary human pDCs isolated from more than 100 bags of human peripheral blood buffy coats. Using the active domain of TLR9 as a bait, we screened the library to identify potential TLR9-interacting proteins in human pDCs activated by CpG-ODN.
Here, we report the identification of phospholipid scramblase 1 (PLSCR1) as a TLR9-binding protein that plays an important role in type 1 IFN-responses in pDCs by regulating TLR9 expression and trafficking.
Results
Interaction of PLSCR1 with TLR9
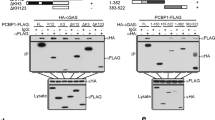
To identify TLR9-interacting proteins in primary human pDCs activated by CpG-ODN, we performed a yeast two-hybrid screen of human pDC cDNA expression library using amino acids 478-768 of the LRR domain of TLR9 as a bait (Figure 1A)7. We identified PLSCR1 by DNA sequencing of 269 clones that were selected by culturing in media deprived of AHLT (adenosine, histidine, leucine, tryptophan) and the ability to metabolize X-gal (Figure 1B). The ability of PLSCR1 to bind TLR9 was further confirmed by co-immunoprecipitation experiments using whole-cell lysates prepared from HEK-293T cells expressing HA-TLR9 and Myc-PLSCR1 (Figure 1C). To investigate whether endogenous PLSCR1 and TLR9 interact with each other, we performed co-immunoprecipitation experiments using whole-cell lysates from the human pDC cell line GEN2.2 that was shown to display similar phenotype and function of human primary pDCs19 with an anti-PLSCR1 antibody or control IgG, followed by immunoblotting with anti-TLR9 antibody. We found that PLSCR1 binds to TLR9 endogenously (Figure 1D). Although many studies found PLSCR1 not only in the plasma membrane (palmitoylated) but also in endosome, golgi and nucleus (unpalmitoylated), it is likely that the fraction of non-membrane-bound PLSCR1 binds to TLR920.

PLSCR1 binds to TLR9. (A) Schematic diagram of human TLR9 shows its different domains. EC, extracellular, TM, transmembrane, and CP, denote cytoplasmic domains. Amino acids 478-768 of TLR9 were used as a bait for yeast two-hybrid screen. (B) Clone 32 with other clones regrew in more stringent condition (media lacking adenine, histidine, leucine, and tryptophan (AHLT) without X-gal, or, with X-gal (LT). (C) Reciprocal interaction of Myc-PLSCR1 and HA-TLR9. Lysates from over-expressing PLSCR1, TLR9 in HEK-293T cells were co-immunoprecipitated with anti-Myc or anti-HA or IgG (control) and western blot was performed (upper two panels). The lower two panels show similar bands on the same blot when reprobed with anti-PLSCR1 (bottom) and anti-TLR9 (upper) antibodies. (D) Endogenously expressed TLR9 binds to PLSCR1. GEN2.2 cells were lysed, precleared, and immunoprecipitated with anti-PLSCR1 antibody or IgG and western blot was performed with anti-TLR9 antibody. Lower panel shows expression of PLSCR1 and upper panel shows the binding to TLR9. These data are representative of three independent experiments.
N-terminal G-box domain of PLSCR1 binds to TLR9
PLSCR1 has the following domains: a G-box-binding domain (aa 3-157) responsible for protein-protein interaction, which overlaps with the N-terminal proline-rich domain (aa 13-97) that binds to SH3 and WW-motif-containing proteins; a DNA-binding domain (aa 86-118); a scramblase domain (aa 86-307) that facilitates trans-bi-layer lipid movement; a nuclear localization signal (aa 257-266); two tyrosine phosphorylation sites (Y69 and Y74); a palmitoylation site (aa 184-189) required for its localization; a trans-membrane domain (aa 291-309); and a 9-aa C-terminal extracellular domain. To determine the binding site of PLSCR1 to TLR9, different forms of PLSCR1 deletion and point mutation mutants were made in a Myc-pCMV expression vector (Figure 2A); each construct was co-transfected with an HA-TLR9 expression vector into HEK-293T cells. Co-immunoprecipitation experiments were performed using whole-cell lysates prepared from the transfected HEK-293T cells. As shown in Figure 2B (left side), the PLSCR1 deletion mutant containing only the N-terminal G-box-binding domain of PLSCR1 (Figure 2A, aa 1-157) could bind to TLR9, whereas the PLSCR1 deletion mutant only containing the C-terminal scramblase domain (Figure 2A, aa 86-307) did not bind to TLR9. This indicates that the proline-rich region present between aa 1-85 of PLSCR1 may be responsible for binding to TLR9. The experiments were repeated using a series of PLSCR1 mutants with N-terminal deletions (Figure 2A), and demonstrated that aa 50-157 within the G-Box domain (aa 1-157) of PLSCR1 was the minimum region necessary for TLR9-binding (Figure 2B, right side). Other mutations at the nuclear localization site, tyrosine phosphorylation sites and the palmitoylation sites of PLSCR1 did not affect the binding to TLR-9.

TLR9 binds to G-Box binding domain of PLSCR1. (A) Schematic presentation of the human PLSCR1 shows the different domains. All the point and deletion mutations were made as described in Materials and Methods. Binding of PLSCR1 to TLR9 is shown on the right side. (B) Full-length, point and deletion mutants of Myc-PLSCR1 plus empty vector were co-transfected with HA-TLR9 into HEK-293T cells. Cell lysates were immunoprecipitated with anti-Myc beads and immunoblots were done with either anti-HA or anti-Myc antibodies.
Binding of PLSCR1 to TLR9 is necessary for type 1 IFN production. When different PLSCR1 fragments were overexpressed in GEN2.2 cells, the scramblase domain alone (unable to bind to TLR9) behaved similar to the control vector, whereas the G-box-binding domain (binds with TLR9) exhibited a similar effect as full-length PLSCR1 (Supplementary information, Figure S1).
PLSCR1 plays an important role in TLR7/9-dependent Type I IFN production
To investigate the function of PLSCR1 in human pDCs, expression of PLSCR1 or MyD88 was knocked down through the use of siRNAs in a human pDC cell line (GEN2.2) that displays a similar phenotype and function to human primary pDCs19. We also used a scrambled siRNA as a control. The efficiency of PLSCR1 knockdown by the siRNA in the pDC cell line was over 90%, as analyzed by western blot (Figure 3A). Control pDCs with scrambled siRNA and pDCs with PLSCR1- or MyD88-specific siRNA were stimulated with CpG-A or CpG-B and their cytokine responses were measured by ELISA. Compared to control pDCs, PLSCR1 knockdown led to over 75% reduction in IFN-α production, about a 20% reduction in IL-6 production and almost a 40% reduction in TNF-α production in response to CpG-A, while MyD88 knockdown led to a more dramatic reduction in IFN-α, IL-6 and TNF-α production in response to CpG-A (Figure 3B). In response to CpG-B, residual IFN-α production was almost completely blocked in pDCs with PLSCR1 or MyD88 knockdown compared to control cells. Meanwhile, MyD88 knockdown led to a dramatic reduction in IL-6 and TNF-α production and PLSCR1 knockdown only marginally reduced IL-6 and TNF-α production by the pDC cell line in response to CpG-B (Figure 3C). These data suggest that PLSCR1 may play a more important role in IFN-α production than in IL-6/TNF-α production in pDCs.

PLSCR1 plays an important role in the IFN-α production in pDC cell line. (A) PLSCR1 knockdown was confirmed by western blot. (B,C) A human pDC cell line were transfected with siRNAs for 48 h followed by stimulation with (B) CpG-A (1 μM) or (C) CpG-B (0.5 μM) for 20 h in 96-well culture plates. IFN-α, TNF-α, and IL-6 production were measured by ELISA. These data are representative of three independent experiments performed in duplicates.
To further investigate the role of PLSCR1 in regulating IFN responses in pDCs, pDCs were generated by obtaining bone marrow (BM) cells from PLSCR1-deficient mice and wild-type control mice and culturing the cells with the Flt3-ligand21 as described in Materials and Methods. The cultured pDCs were stimulated with CpG-A or CpG-B and their cytokine responses were measured by ELISA. We found that pDCs from PLSCR1-deficient mice produced much lower levels of IFN-α and similar levels of IL-6 and TNF-α compared to wild-type pDCs (Figure 4A). These data further confirmed that PLSCR1 may play a more important role in regulating IFN-α, than IL-6 or TNFα production in pDCs. We obtained similar results from the BM-derived pDCs when they were activated by herpes simplex virus 1 (HSV-1) (Figure 4B) or influenza virus A (Flu) (Figure 4C). These data suggest that PLSCR1 plays important roles in type 1 IFN production in pDCs induced by TLR7/9 activation.

PLSCR1 positively regulates TLR7/9-dependent IFN-α production in mice pDCs. pDCs were isolated from mice BM and cultured for seven days with Flt3L. Flt3L-driven pDCs (1 × 105 cells) were treated with 1 μM CpG-A or 0.5 μM CpG-B (A), HSV (B) or Flu (C) (multiplicity of infection, MOI, 10) for 20 h and the concentration of IFN-α, IL-6, and TNF-α production were measured from the supernatants of each condition by ELISA. Horizontal solid lines intersecting circular data points represent means; ** indicates P < 0.0035; *** indicates P< 0.0003; NS indicates no statistical significane.
In addition, we measured type 1 IFN production in isolated BM-derived macrophages and monocyte-derived dendritic cells (mDCs) as well as pDCs from WT and KO mice stimulated by CpG-A, Flu and HSV (Supplementary information, Figure S2). We observed that Flu did not elicit a good response in macrophages but the HSV did and led to a similar pattern as we have seen earlier for pDCs (Figure 4). Similar patterns of responses were observed for mDCs as that for pDCs when stimulated with either CpG-A or Flu.
PLSCR1 recruits TLR9 to the endosome
To investigate whether PLSCR1 colocalizes with TLR9, we performed double-immunofluorescence staining with anti-PLSCR1 (red) and anti-TLR9 (green) in pDCs generated from wild-type mouse BM cells cultured with Flt3L. Figure 5A (upper three panels) shows that PLSCR1 was colocalized with TLR9 in the ER. In addition, PLSCR1 was also found on the cell plasma membrane (Figure 5A, lower panel).

Recruitment of TLR9 to endosomes by PLSCR1. (A) From upper to lower, colocalization of PLSCR1 (red) with TLR9 (green), with ER (green) and ER (green) with TLR9 (red) in wild-type mice BM-pDC (upper panel); expression of PLSCR1 (red) in plasma membrane (PM, green) (lower panel). (B) TLR9 and PLSCR1 in wild-type and PLSCR1-deficient mice BM-pDCs were stimulated with Alexa Flour 488-labeled CpG-A (2 μM) for 5 h and analyzed for TLR9 (red/green), PLSCR1 (red) colocalization with CpG-A (green) in EE using anti-Transferrin receptor 1 as a marker (red). These data are the representative of three independent experiments.
To investigate whether PLSCR1 plays an important role in TLR9 and CpG-DNA trafficking in pDCs, we analyzed the localization of TLR9 and CpG-DNA in the EE in pDCs from wild-type mice and PLSCR1-deficient mice following stimulation with CpG-A. We found that at 5 h following CpG-A stimulation, CpG-A was colocalized with PLSCR1 and TLR9 within the EEs in pDCs from wild-type mice (Figure 5B). However, in pDCs from PLSCR1-deficient mice, only CpG-A-DNA but not TLR9 was found within the EEs. These data suggest that PLSCR1 may play an important role in TLR9 trafficking to the EEs.
PLSCR1 triggers downstream signaling to translocate IRF7
To investigate the requirement of PLSCR1 for CpG-triggered signaling culminating in the activation of IRF7, i.e., translocalization of IRF7 to the nucleus, we isolated cytosolic and nuclear fractions following CpG-A treatments and determined the nuclear translocation of IRF7 (Figure 6A), and performed knockdown of MyD88, TLR9, and PLSCR1 in the human pDC cell line (Figure 6B) and monitored nuclear localization of IRF7 (Figure 6C). When MyD88 or TLR9 expression was knocked down by siRNA, nuclear localization of IRF7 by CpG-A stimulation was diminished. Interestingly, knockdown of PLSCR1 also diminished the nuclear localization of IRF7 in cells treated with CpG-A. We also found that in PLSCR1 KO mice, the nuclear translocalization of IRF7 is hampered upon stimulation by CpG-A (Figure 6D). These results suggest that PLSCR1-mediated TLR9 signaling is necessary for nuclear translocation of IRF7.

PLSCR1 triggers downstream signaling cascades. (A) Nuclear fractions from pDC cell line were stimulated with 2 μM of CpG-A for the indicated times and were immunoblotted with anti-IRF7 antibody. HDAC1 and β-actin were used as nuclear and cytosolic markers, respectively. (B) Endogenous expression of PLSCR1, TLR9, and MyD88 were monitored by immunoblotting with anti-PLSCR1, anti-TLR9, and anti-MyD88 antibodies, as indicated. (C) Nuclear fractions from GEN2.2 cells transfected with siRNA, as indicated, upon treatment with 2 μM of CpG-A for 3 h were immunoblotted with anti-IRF7 antibody. HDAC1 was used as a loading control. (D) Colocalization of IRF7 with the nucleus in wild-type and PLSCR1-deficient BM-pDCs treated with CpG-A for 5 h.
Discussion
pDCs are a specialized cell type in the innate immune system that uses TLR7/9 in the endosomes to sense viral nucleic acids. Although genetic approaches have identified several molecules such as gp96, UNC93B1, AP3, and Viperin22 that play key roles in TLR9-mediated DNA-sensing and type 1 IFN responses, the overall nature of TLR9-interacting proteins in primary pDCs remained to be fully understood. Technically, it has been very challenging to directly study protein-protein interaction in pDCs because of the difficulty in obtaining a large number of pDCs from human and mouse tissues or tissue cultures. To overcome this technical difficulty, we have constructed a yeast two-hybrid cDNA expression library from 100 million human primary pDCs. Using the active domain of TLR9 as a bait, we have identified a number of potential TLR9-interacting proteins. In this study, we show that PLSCR1 is a TLR9-binding protein by co-IP experiments. Results from co-IP experiments using serial deletions of PLSCR1 further show that the proline-rich G-box-binding domain of PLSCR1 binds to TLR9. By siRNA knockdown experiments, we show that PLSCR1 plays a key role in TLR9-mediated signaling and type 1 IFN responses induced by CpG-ODN. This was confirmed by experiments using pDCs from PLSCR1-deficient mice. By using confocal microscopy, we show that the expression level of TLR9 and the translocation of TLR9 from ER to endosomes were compromised in PLSCR1-deficient pDCs. This result suggests that PLSCR1 is involved in TLR9 trafficking from the ER to the endosome. PLSCR1 was isolated as a TLR9-interacting protein from a human pDC library using part of the LRR domain of TLR9 as a bait, a position at which TLR9 is cleaved to generate the functional C-terminal TLR9 that binds to ligands and MyD887,11. However, PLSCR1 could also bind the full-length TLR9 in pDCs. This suggests that the association of PLSCR1 with TLR9 occurs before entering into the endosome and this association does not require the cleavage of TLR9. Indeed, we found normal TLR9 cleavage in pDCs derived from PLSCR1-deficient mice (data not shown).
PLSCR1 is a 35 kd protein that plays a key role in the ATP-independent bidirectional trans-bilayer migration of phospholipids upon binding of calcium ions that results in a loss of phospholipid asymmetry in the plasma membrane23. Several studies have shown that PLSCR1 is a type 1 IFN-inducible gene24. Other studies have shown that PLSCR1 is important for anti-viral immune responses because PLSCR1-deficient mice are susceptible to VSV infection, and PLSCR1 is critical for the expression of many IFN-inducible genes during viral infection25. Increased expression of PLSCR1 was found in monocytes from patients with systemic lupus erythematosus26. These data suggest that PLSCR1 may play an important role in type 1 IFN responses during anti-viral innate immunity or in sensing self-DNA/RNA in autoimmune diseases. Our study provides a molecular explanation on the key role of PLSCR1 in TLR9-mediated type 1 IFN-responses in pDCs. The identification of PLSCR1 as a TLR9-interacting protein represents a very important step towards fully understanding the TLR9-signaling pathway, which is critical for anti-viral innate immune responses and self-nucleic acids-associated autoimmune diseases.
Materials and Methods
Reagents and antibodies
Synthetic ODNs (C-phosphate-G (CpG)-A, ODN2216; CpG-B, ODN2006) were purchased from Sigma. The following antibodies were used for immunoblotting: anti-myeloid differentiation primary response gene 88 (anti-MyD88) (eBioscience), anti-Toll-like receptor9 (anti-TLR9) (eBioscience), anti-IRF7, anti-PLSCR1 (Santa Cruz Biotech.) anti-HA-HRP, and anti-β-actin (Sigma). ELISA kits for human IFN-α (Bender MedSystems), human TNF-α, and IL-6 (R & D Systems) were used. A nuclear extraction kit (Pierce) was used to investigate nuclear extraction of IRF7.
Plasmids construction and screening of the yeast two-hybrid library
The LRR domain of TLR-9 (LRR-15 to LRR-26, amino acids 478-768) was digested at the EcoRI and SalI sites and ligated to the pGBKT7 vector at the same sites, which expresses proteins fused to amino acids 1-147 of the GAL4-DNA-binding domain (DNA-BD) (Clontech). This fragment of TLR9 was used as bait to screen a human pDC cDNA library fused to the GAL4 activation domain according to the manufacturer's (Clontech) instructions. Positive clones were verified by one-on-one transformations and selection on agar plates lacking adenine, histidine, leucine, and tryptophan (AHLT) and also processed for α-galactosidase assay. A total of 269 clones were purified and sequenced at the University of Texas M.D. Anderson DNA Sequencing Core Facility. All the human PLSCR1 constructs were made by cutting and ligating at EcoRI and NotI sites into pCMV-Myc (Clontech) expression vector. Primers were used for all the constructs made in HA- and Myc-vectors are shown in Table 1.
Plscr1-knockout mice
The generation of Plscr1-knockout mice was described previously27. Heterozygous Plscr1 (Plscr1+/−) mice in a 129/SvEvBrd genetic background were produced and obtained from the Texas Institute for Genomic Medicine (TIGM, www.tigm.org). Targeting was performed using a genomic DNA fragment containing exons 4-8 of murine PLSCR1 (representing amino acid sequence Pro33 to Asp310, Genbank accession no. AF159593) inserted in the vector pKOS, using 129/SvEvBrd embryonic stem cells. Targeted clones with confirmed deletion of the Plscr1 gene locus were injected into 129/SvEvBrd blastocysts, and germ-line transmission was confirmed. Genotyping was performed by PCR of tail genomic DNA according to manufacturer's procedures. Animals with targeted disruption of the Plscr1 gene were obtained and back-crossed more than five times with C57BL/6 mice. Animals were housed in pathogen-free barrier facilities. All experiments reported here were performed using mice and cells derived from the inbred C57BL/6 strain and according to the institutional guidelines at The University of Texas MD Anderson Cancer Center.
Isolation, in vitro culture and antibody staining of mouse pDCs
BM cells were isolated as described previously25 from mice by flushing femurs and tibiae with RPMI supplemented with 10% heat-inactivated FCS. The cells were then passed through a 70-μm cell strainer, centrifuged, and resuspended in a Tris-ammonium chloride buffer (Sigma-Aldrich) at 37 °C for 5 min to lyse the red blood cells. Intact cells were centrifuged and resuspended at a concentration 2 × 106 cells/ml in culture medium consisting of RPMI 1640, 10% FCS, 1 mM sodium pyruvate, HEPES, penicillin, streptomycin, and 2-mercaptoethanol supplemented with murine Flt3L (100 ng/ml; R&D Systems). Every fourth day of culture, half of the medium was removed and was replaced with fresh cytokine-supplemented culture medium. BM cells were cultured in the presence of Flt3L (100 ng/ml, R&D Systems) for 7 days. Negative immuno-selection of lineage-negative bone-marrow progenitors was performed with biotinylated anti-CD3, anti-CD11b, anti-CD19, and anti-Ter119 Abs, followed by SA-coated micro beads and were recovered by magnetic separation (Miltenyi Biotec). Cells were stained with anti-B220-FITC, anti-CD11c-PE, anti-CD11b-APC mAb, and CD11c+CD11b-B220+ were sorted for pDCs by fluorescence-activated cell sorting on a FACS Aria machine (BD Biosciences).
In vitro stimulation and quantification of cytokine production
Freshly isolated mouse pDCs from BM were re-suspended in culture medium (4 × 104 cells/100 μl) and stimulated with 1 μM CpG-A or 0.5 μM CpG-B, HSV-1, Flu (multiplicity of infection, MOI, 10) for 20 h. For quantification of cytokine production, cell-free supernatants were collected after 20 h and analyzed by ELISA (R&D systems).
Cell lines, tissue cultures, plasmids, and transfection
HEK293T cells were purchased from American Type Culture Collection and cultured in RPMI (GIBCO) supplemented with 10% FBS (Atlanta), L-glutamine, penicillin/streptomycin, and sodium pyruvate (GIBCO). GEN2.2 cells were cultured in GlutaMax-RPMI (GIBCO) supplemented with 10% FBS, MEM-nonessential amino-acid solution (GIBCO), penicillin/streptomycin, and sodium pyruvate. Transfection of GEN2.2 cells was performed using an electroporation-based transfection solution V-kit (Amaxa) with 5 × 106 cells and 0.4 nmol of siRNA. siPLSCR1 (Dharmacon) and siTLR9 (Sigma-Aldrich), and other siRNAs were purchased from Dharmacon. The program number for electroporation was A033. The sequences of siRNAs are as follows: siTLR9, 5′-CUUAACCUGUCCUUCAAUU-3′. The sequences of siMyD88 pool are as follows: siMyD88-1, 5′-CGACUGAAGUUGUGUGUGU-3′; siMyD88-2, 5′-GCUAGUGAGCUCAUCGAAA-3′; siMyD88-3, 5′-GCAUAUGCCUGAGCGUUUC-3′; siMyD88-4, 5′-GCAGGUGUGUCUGGUCUAU-3′. siControl is a non-targeting pool from Dharmacon.
Measurement of cytokine production from GEN2.2 cells
GEN2.2 cells transfected with siRNAs were stimulated with 1 μM of CpG-A or 0.5 μM CpG-B for 20 h. Concentrations of IFN-α, TNF-α, and IL-6 in the culture supernatants were measured by ELISA in duplicate for a total of three times.
In vitro pull-down and immunoblotting assays
Lysates from HEK-293T cells co-transfected with expression plasmids encoding a full-length or truncated Myc-PLSCR1 and a full-length HA-TLR9 or control vectors was incubated with anti-HA and anti-Myc monoclonal antibodies for 4 h or overnight. Bound HA-tag and Myc-tag proteins were detected by immunoblotting with anti-HA-HRP antibody and anti-Myc-HRP antibodies.
Immunofluorescence analysis
The purified pDCs were left untreated or were treated with CpGs for 5 h and then were placed onto cover-slips pretreated with poly-D-lysine, fixed with 4% paraformaldehyde, and permeabilized with Permeabilization Reagent B (Invitrogen). After the coverslips were blocked with 10% secondary antibody, species-specific serum and 0.1% BSA in PBS, the pDCs were probed with primary antibodies (anti-TLR9, anti-PLSCR1, anti-cholera toxin unit B, anti-TfR-biotin overnight at 4 °C), and alexa flour 488-conjugated CpG-A. After incubation for 30 min with secondary antibodies (Invitrogen, Alexa Fluor 647 donkey anti-mouse IgG1 for anti-TLR9, Alexa Fluor 555 goat anti-mouse IgG1 for anti-PLSCR1, and Alexa Fluor 405 streptavidin for anti-TfR-biotin), the coverslips were mounted with Prolong Gold (Molecular probes) with or without DAPI.
Statistical analysis
Statistically significant differences were determined by unpaired, two-tailed, Student's t-test. P-values < 0.05 were considered statistically significant. Statistical analysis of data was done using GraphPad Prism version 5 for Macintosh (GraphPad Software).
References
Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999; 284:1835–1837.
Cella M, Jarrossay D, Facchetti F, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nature Med 1999; 5:919–923.
Gilliet M, Cao W, Liu YJ . Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nature Rev Immunol 2008; 8:594–606.
Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM . The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol 2007; 177:265–275.
Kim YM, Brinkmann MM, Paquet ME, Ploegh HL . UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008; 452:234–238.
Yang Y, Liu B, Dai J, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 2007; 26:215–226.
Ewald SE, Lee BL, Lau L, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 2008; 456:658–662.
Chockalingam A, Brooks JC, Cameron JL, Blum LK, Leifer CA . TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol Cell Biol 2009; 87:209–217.
Sasai M, Linehan MM, Iwasaki A . Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 2010; 329:1530–1534.
Latz E, Schoenemeyer A, Visintin A, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 2004; 5:190–198.
Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL . Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol 2008; 9:1407–1414.
Häcker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H . Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med 2000; 192:595–600.
Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408:740–745.
Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005; 434:772–777.
Takaoka A, Yanai H, Kondo S, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 2005; 434:243–249.
Kerkmann M, Costa LT, Richter C, et al. Spontaneous formation of nucleic acid-based nanoparticles is responsible for high interferon-alpha induction by CpG-A in plasmacytoid dendritic cells. J Biol Chem 2005; 280:8086–8093.
Guiducci C, Ott G, Chan JH, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med 2006; 8:1999–2008.
Honda K, Ohba Y, Yanai H, et al. Spatiotemporal regulation of MyD88–IRF7 signalling for robust type I interferon induction. Nature 2005; 434:1035–1040.
Chaperot L, Perrot I, Jacob MC, et al. Leukemic plasmacytoid dendritic cells share phenotypic and functional features with their normal counterparts. Eur J Immunol 2004; 34:418–426.
Wiedmer T, Zhao J, Nanjundan M, et al. Palmitoylation of phospholipid scramblase 1 controls its distribution between nucleus and plasma membrane. Biochemistry 2003; 42:1227–1233.
Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS . The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 2008; 28:509–520.
Saitoh T, Satoh T, Yamamoto N, et al. Antiviral protein viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 2011; 34:352–363.
Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ . Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem 1997; 272:18240–18244.
Zhou Q, Zhao J, Al-Zoghaibi F, et al. Transcriptional control of the human plasma membrane phospholipid scramblase 1 gene is mediated by interferon-alpha. Blood 2000; 95:2593–2599.
Dong B, Zhou Q, Zhao J, et al. Phospholipid scramblase 1 potentiates the antiviral activity of interferon. J Virol 2004; 78: 8983–8993.
Suzuki E, Amengual O, Atsumi T, et al. Increased expression of phospholipid scramblase 1 in monocytes from patients with systemic lupus erythematosus. J Rheumatol 2010; 37:1639–1645.
Zhou Q, Zhao J, Wiedmer T, Sims PJ . Normal hemostasis but defective hematopoietic response to growth factors in mice deficient in phospholipid scramblase 1. Blood 2002; 99: 4030–4038.
Acknowledgements
This work was supported by a grant from the National Institutes of Health awarded to YJL (U19 AI071130). We thank David He and Karen Ramirez for assistance with cell sorting. The FACS Core Facility is supported by the M. D. Anderson Cancer Foundation and in part by the National Institutes of Health through M. D. Anderson Cancer Center Support Grant CA016672. GEN2.2 cells were obtained through a Material Transfer agreement with Dr Joel Plumas and Lawrence Chaperot from the Université Joseph Fourier, and INSERM U823, France. We also acknowledge Melissa Wentz for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
PLSCR1 and TLR9 interaction is necessary for IFN-α production. (PDF 47 kb)
Supplementary information, Figure S2
Responses of TLR9 and TLR7/8 pathway in macrophages and in mDC. (PDF 61 kb)
Rights and permissions
About this article
Cite this article
Talukder, A., Bao, M., Kim, T. et al. Phospholipid Scramblase 1 regulates Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Cell Res 22, 1129–1139 (2012). https://doi.org/10.1038/cr.2012.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2012.45
Keywords
This article is cited by
-
Phospholipid scramblase 1: a protein with multiple functions via multiple molecular interactors
Cell Communication and Signaling (2022)
-
Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function
Nature Communications (2018)
-
BAD-LAMP controls TLR9 trafficking and signalling in human plasmacytoid dendritic cells
Nature Communications (2017)
-
The multifaceted biology of plasmacytoid dendritic cells
Nature Reviews Immunology (2015)
-
Regulation of TLR7/9 signaling in plasmacytoid dendritic cells
Protein & Cell (2013)
