
Abstract
To deal with the constant challenge of protein misfolding in the endoplasmic reticulum (ER), eukaryotic cells have evolved an ER protein quality control (ERQC) mechanism that is integrated with an adaptive stress response. The ERQC pathway is comprised of factors residing in the ER lumen that function in the identification and retention of aberrantly folded proteins, factors in the ER membrane for retrotranslocation of misfolded polypeptides, and enzymes in the cytosol that degrade retrotranslocated proteins. The integrated stress response (termed ER stress or unfolded protein response, UPR) contains several signaling branches elicited from the ER membrane, which fine-tune the rate of protein synthesis and entry into the ER to match the ER folding capacity. The fitness of the cell, particularly those bearing a high secretory burden, is critically dependent on functional integrity of the ER, which in turn relies on these stress-attenuating mechanisms to maintain protein homeostasis, or proteostasis. Aberrant proteostasis can trigger cellular apoptosis, making these adaptive stress response systems attractive targets for perturbation in treatment of cell malignancies. Here, we review our current understanding of how the cell preserves ER proteostasis and discuss how we may harness the mechanistic information on this process to develop new cancer therapeutics.
Similar content being viewed by others
Introduction
Secretory and membrane proteins begin their journey in the cell when they are translocated into the lumen of the endoplasmic reticulum (ER) or integrated into the ER membrane via the Sec61 translocon. An elaborate cohort of chaperones in the ER then assist polypeptides in folding and assembly so that they acquire functional, native conformations, as well as proper oligomeric states. Correctly folded and assembled polypeptides are then sorted to ER exit sites where they are loaded into distinct vesicles for transport to different cellular destinations. Successful completion of this long journey is not a trivial task. Polypeptides must pass stringent check points enforced by an evolutionarily conserved ER protein quality control program (ERQC), which efficiently targets and destroys aberrant polypeptides or unassembled protein complexes 1. The accumulation of aggregation-prone misfolded polypeptides is so detrimental to the fitness of the cell that the ERQC has evolved to destroy almost any protein of questionable quality, such as those that stay in the folding process for a prolonged period of time.
The cell is constantly modulating its protein folding and degradation capacities to avoid accumulation of misfolded proteins and maintain protein homeostasis, or proteostasis. When the production of misfolded proteins exceeds degradation, as often occurs in damaged or aging cells, or in cells exposed to chemical agents that perturb protein folding or the ERQC pathway, the unfolded protein response (UPR) is elicited. The UPR can mobilize several additional mechanisms to restore ER homeostasis 2, 3. However, if these efforts fail to overcome the folding crisis, persistent ER stress can switch on an apoptotic program, which results in cell elimination 4.
Cells with different specialized functions can bear different secretory burdens, resulting in different levels of intrinsic stress among various cell types. Consequently, cells can have distinct sensitivities to extra stressors that perturb ER proteostasis. For example, the plasma B cells are specialized for antibody production. To accommodate this increased secretory burden, the ER in differentiated plasma B cells is drastically expanded to boost the folding capacity. Despite this adaptive change, the massive secretory flux still puts these cells at risk for apoptosis induced by external stressors that might otherwise be inert. Indeed, a recent study demonstrated that plasmacytic differentiation without increased secretory load confers resistance to the proteasome inhibitor bortezomib 5. Thus, specific therapeutic strategies aimed at targeting ER proteostasis may be employed to selectively treat certain tumors carrying a high secretory burden 6, 7.
ER-associated protein degradation (ERAD)
Elimination of misfolded proteins from the ER by the ERQC program counteracts the production of aberrant proteins from various folding mishaps. This essential housekeeping function operates in the ER of all eukaryotic cells. Misfolded proteins are exported from the ER and subsequently destroyed by the ubiquitin-proteasome system in the cytosol by a process called retrotranslocation or ER-associated protein degradation (ERAD) 8, 9. In this section, we will discuss the players identified to date in this pathway based on their known functions in order to reveal potential sites of perturbation for targeted cancer therapy.
Substrate recognition and targeting
Misfolding signals can reside in a variety of locations on a polypeptide. Nonetheless, all such signals seem to be associated with the exposure of certain hydrophobic patches that are normally embedded in the interior of a folded polypeptide or properly assembled protein complex. Misfolding signals on polypeptides are usually recognized by chaperones 10, 11. For secretory and ER luminal proteins, misfolding signals reside entirely within the ER and thus must be recognized as such in the ER lumen. For membrane proteins, depending on their topology and the position of the lesions, substrate recognition may occur in the ER lumen, within the membrane, or in the cytoplasm. Accordingly, studies in yeast have classified ERAD substrates into three categories, called ERAD-L, ERAD-M, and ERAD-C based on the location of the lesions in the lumen, membrane, or cytosol, respectively 12, 13. Each substrate cohort can also be distinguished by its unique genetic and biochemical requirements for degradation 12, 14. For example, the degradation of the ERAD-L and ERAD-M substrates is strictly dependent on the ubiquitin ligase Hrd1p (see below), whereas ERAD-C substrates require a different ligase Doa10p for turnover 12, 15. In mammalian cells, the degradation of different categories of ERAD substrates does not seem to follow the exact same pattern of labor division 16. Perhaps, the increased complexity of the secretory pathway in these cells calls for more flexibility and redundancy in the ERQC program.
The rules for substrate recognition by ER chaperones can be quite degenerative. In some cases, chaperones act in parallel 17, whereas other times they may work sequentially to 'interrogate' substrates bearing multiple misfolding signals 18, 19. In general, a short hydrophobic segment exposed on a misfolded protein can be recognized by the Hsp70 family of chaperones such as glucose regulated protein of 78kDa (GRP78/BiP) in mammals and Kar2p in yeast 20, 21, which may be sufficient to initiate retrotranslocation. However, it is worth mentioning that the Hsp70 family of chaperones normally functions as a folding catalyst. BiP/GRP78 also associates with the Sec61 complex and can act as a ratcheting molecule in this context to facilitate the translocation of polypeptides into the ER 22. Thus, the early steps of the ERQC pathway overlap with ER protein biogenesis. It is currently unclear how chaperones can switch their job from a folding assistant to an ERQC triaging factor. One possibility is that prolonged association of a polypeptide with one or more chaperones without productive folding is sufficient to target the chaperone-substrate complex to a retrotranslocation channel (see below) in the ER membrane, which triggers export and destruction. An analogous timer mechanism has been proposed for the disposal of misfolded glycoproteins (Figure 1).

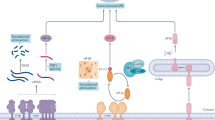
The proposed timer mechanism for degradation of misfolded glycoproteins. Upon entry into the ER, glycoproteins are folded with the assistance of the lectins, Calnexin and Calreticulin. In the so-called Calnexin cycle, the terminal glucose residues on a glycan are removed by glucosidases I and II. In higher eukaryotic cells, a UDP-glucose:glycoprotein glucosyltransferase (UGGT) can subsequently add a glucose residue back if the protein is unfolded. The mono-glucose residue is recognized by Calnexin and Calreticulin, which retains unfolded proteins in the folding cycle. The deglucosylated proteins can exit the Calnexin cycle upon folding, or when the mannose in the N-glycan is cleaved by the ER mannosidase I (Man I). Cleavage of mannose irreversibly extracts glycoproteins out of the folding cycle, which is followed by further mannose trimming by Htm1p/EDEM. Neither mannosidase I nor Htm1p/EDEM displays strong activities in vitro (as indicated by the thin arrows). Perhaps, the sluggish action of these enzymes gives newly synthesized glycoproteins sufficient time to fold in the Calnexin cycle. Mannose trimming by ER Man I and Htm1p/EDEM generates a glycan signal that is recognized by the downstream lectin Yos9. Yos9 resides in a large membrane protein complex containing other ERAD factors, which initiates retrotranslocation.
A large number of proteins entering into the ER carry the consensus sequence Asn-X-Ser/Thr (X designates any residues) that can be modified with an N-linked oligosaccharide (GlcNAc2-Man9-Glu3), which adds an extra layer of complexity for substrate recognition in the ERQC. Glycoproteins are usually folded with the assistance of the lectins calnexin and calreticulin, the glucosidase-1 and -2, and the UDP-glucose:glycoprotein glucosyltransferase (UGGT) (Figure 1) 23, 24. If a polypeptide remains unfolded after extended association with these lectins, ER-resident mannosidases (Htm1p in yeast or EDEM in mammals) associated with calnexin may trim mannose from the glycan 25, 26, 27, generating a signal containing a terminal α1, 6-linked mannose residue 28, 29, 30, which is then recognized by the downstream lectin Os9/Yos9p 31, 32. This results in the extraction of the polypeptide from the folding pathway, which causes its retrotranslocation and degradation. Interestingly, Yos9p not only recognizes the trimmed glycan using its mannose 6-phosphate receptor homology (MRH) motif, but it also has the capacity to recognize unfolded proteins 33, 34, presumably via association with an exposed hydrophobic patch. Accordingly, the number and the position of glycans relative to the misfolding signal in a glycoprotein can influence the rate of substrate recognition and degradation 35.
Protein folding in the ER often involves the formation of disulfide bonds, which is catalyzed by the family of Protein Disulfide Isomerases (PDIs). PDI also has the capacity to break non-native disulfide bonds and reshuffle them to form proper disulfide links. Like other folding catalysts mentioned above, PDI can also partake in the ERQC program by facilitating substrate recognition and partition in the ERAD pathway. Several lines of evidence suggest that the ERAD machinery has limited capacity when handling tightly folded substrates or protein aggregates containing intermolecular disulfide bonds. Bhamidipati and colleagues demonstrated that the fusion of a tightly folded domain to the ERAD-L substrate CPY* impedes its degradation 36. During retrotranslocation, polypeptides presumably move across the ER membrane in an unfolded monomeric form. In line with this notion, a recent study shows that two basic intramembrane residues in unassembled TCRα chain are required to prevent TCRα oligomerization via inter-chain disulfide bond formation, and this facilitates its degradation 37. Moreover, Okuda-Shimizu et al. found that retrotranslocation of non-secreted κ-LC involves the conversion of a partially oxidized precursor into a reduced monomer 38. Thus, PDI may promote retrotranslocation by breaking disulfide bonds in oligomerized ERAD substrates, which generates a retrotranslocation competent monomer. Additionally, PDI was also found to interact with certain retrotranslocation complexes in the ER membrane 39, 40, which may define the point of no return at which substrates are irreversibly switched to the degradation-bound track. Ushioda et al. recently uncovered another ER reductase ERdj5, which may act analogously to PDI to facilitate retrotranslocation. ERdj5 associates with EDEM, which provides a convenient means to couple protein unfolding with mannose trimming and substrate hand-off to a Yos9p-containing retrotranslocation complex in the ER membrane 41.
Retrotranslocation across the ER membrane
Once segregated from the cohort of properly folded polypeptides, misfolded proteins are then bound to the cytosol for degradation by the proteasome. The transit of polypeptides across the ER membrane most likely occurs via a protein conducting channel(s), although a lipid-mediated dislocation hypothesis has also been proposed 42. Early evidence indicated that the Sec61 complex, which mediates polypeptide import into the ER, might also act to translocate misfolded polypeptides out of the ER. Although some genetic and biochemical evidence supports this view 43, 44, 45, the degradation of many ERAD substrates proceeds normally in the absence of a functional Sec61 complex 46, 47, 48, 49. Thus, it seems that the Sec61 complex is unlikely to serve as a major channel for retrotranslocation.
Recent studies suggest that the multispanning membrane proteins Derlin-1, -2 and -3, which reside in a large protein complex together with the ER- associated E3 ubiquitin ligase Hrd1, define a site of retrotranslocation in the ER membrane 50, 51, 52, 53. With four predicted transmembrane segments, the Derlin proteins may serve as a channel component or scaffold/accessory factors that facilitate channel assembly. Intriguingly, Derlins can associate with luminal ERAD factors such as EDEM as well as with the cytosolic ERAD components including the AAA ATPase p97 and the cytosolic N-glycanase. This may provide a physical link between substrate recognition in the ER and dislocation to the cytosol 53, 54, 55. However, definitive evidence in support of a Derlin-containing proteinous channel is still missing. Likewise, the E3 ligase (see below) Hrd1 can also coordinate actions on both sides of the ER membrane through protein-protein interactions 33, 56. In yeast, Hrd1p can interact with ER luminal proteins such as the lectin Yos9p through its interacting partner Hrd3p. It also binds p97/Cdc48p (see below) in the cytosol. Moreover, although the N-terminal transmembrane segments of Hrd1p are not required for its ligase activity 57, they are essential for ERAD function 58. One likely scenario is that the transmembrane domains of Hrd1 may participate in the formation of some kind of membrane pore to translocate ERAD substrates. Consistent with this view, a recent study used an elegant crosslinking approach to demonstrate that a retrotranslocation intermediate is in close proximity to Hrd1p 59.
It is worth mentioning that many ERAD substrates do not require the Derlin-Hrd1 complex for degradation. It is clear from studies in yeast that the multiple-spanning E3 ubiquitin ligase Doa10p acts in parallel to Hrd1p to degrade membrane proteins containing a misfolded cytosolic domain 12, 15. Like Hrd1p, Doa10p also contains multiple transmembrane segments, which may act analogously to Hrd1p in assisting client transport across the ER membrane. Finally, in mammalian cells, newly synthesized major histocompatibility complex (MHC) class I heavy chain can be dislocated from the ER membrane under the influence of the human cytomegaloviral protein US2 by a novel ERAD mechanism that involves signal peptide peptidase and the multiple membrane-spanning ubiquitin ligase TRC8 60, 61. Whether this mechanism is used to degrade naturally occurring misfolded proteins is unclear. Taken all together, the existing evidence suggests the presence of multiple parallel retrotranslocation pathways, each of which channels a subset of misfolded proteins into the cytosol.
The ubiquitination machinery
Once emerging from the ER lumen, polypeptides undergo ubiquitination with one or more chains of ubiquitin molecules covalently linked to either lysine or serine/threonine residues in the substrate 62, 63. Polyubiquitination of ERAD substrates is not only required for subsequent dislocation from the ER membrane, but also for targeting dislocated polypeptides to the 26S proteasome for degradation 64, 65, 66.
Ubiquitination requires the sequential actions of three types of enzymes, an E1 activating enzyme, an E2 conjugating enzyme and an E3 ubiquitin ligase. The human genome contains two E1 enzymes, ∼40 E2s and 600-1 000 E3 ligases. In yeast, the relevant ERAD E2s are Ubc7p, Ubc6p, and Ubc1p 57, 64, 67, 68. Deletion of these genes individually or in combination inhibits ubiquitination and degradation of many misfolded ER proteins. In mammals, the Ube2g and Ube2j sub-families are homologous to the E2s Ubc7p and Ubc6p, respectively 69. Members of these E2 families have been implicated in ERAD of many misfolded substrates 70, 71, 72, 73, 74. In yeast, Hrd1p and Doa10p are the two major ubiquitin ligases dedicated to ERAD 15, 57. In mammals, orthologs of the yeast Hrd1p and Doa10p, as well as a Hrd1-related E3 named gp78, mediate the degradation of most misfolded ER proteins 73, 74, 75, 76, 77, 78, 79. Mammalian cells also employ several additional E3 ligases including RMA1 80, the U-box containing ubiquitin ligase CHIP 81, and Parkin, a RING finger E3 linked to the juvenile Parkinson's disease 82. Two F-box-containing proteins, Fbs1 and Fbs2, each of which is part of a multi-subunit Skp, Cullin, F-box-containing (SCF) ubiquitin ligase, have been shown to recognize carbohydrate chains to facilitate ubiquitination and degradation of retrotranslocated glycoproteins 83, 84. Finally, a ubiquitin chain-elongating factor named Ufd2p in yeast or E4a and E4b in mammals was shown to extend short ubiquitin chains to enhance the efficiency of substrate targeting to the proteasome 85, 86, 87. The involvement of multiple E3 ligases across species in ERAD is consistent with the notion that ubiquitin ligases confer substrate specificity. However, the number of misfolded ER proteins clearly exceeds the available ligases. It is likely that each retrotranslocation complex contains at least one E3 ubiquitin ligase that modify any substrate emerging from the same translocon.
As expected, many ERAD-specific E2 and E3 enzymes are bound to the ER membrane with their catalytic domains facing the cytosol. Ubc6p and the Ube2j family of E2s contain a short carboxyl terminal transmembrane segment that anchors them to the ER membrane. Hrd1, gp78, RMA1, and Doa10 are multi-spanning membrane proteins carrying a cytosolically localized RING-finger domain. The transmembrane segments of these E3s may not only anchor the ligases in the ER membrane, but may also serve a direct role in the retrotranslocation process (see above). Some E2 and E3s such as Ubc7p, CHIP, Parkin, Fbs1 and Fbs2 are soluble proteins. Nonetheless, in yeast, Ubc7p is recruited to the ER membrane by association with the membrane protein Cue1p 65, and in mammals, Ube2g2 interacts with the multispanning membrane ligase gp78 with a high affinity 88, 89. It is currently unclear whether or not the other soluble ubiquitin ligases mentioned above are recruited to the site of translocation by interaction with an ER membrane partner.
The dislocation-driving ATPase Cdc48/p97
Misfolded ER proteins undergoing retrotranslocation are dislocated from the ER membrane before being targeted to the proteasome. The precise mechanism by which polypeptides are extracted from the membrane is unclear, but the major player of this reaction has been identified as the AAA (ATPase associated with various cellular activities) ATPase p97 in mammals or its homolog Cdc48p in yeast 90, 91, 92, 93.
p97 belongs to the type II AAA ATPase family as it contains two similar Walker type AAA ATPase domains. It forms an evolutionarily conserved hexameric ring with a central pore 94, 95. It was proposed that Cdc48/p97 might function as a ubiquitin-selective chaperone to segregate ubiquitinated substrates from a large, relatively immobile entity such as the ER membrane 96. Through its actions on diverse substrates, p97 can influence a variety of cellular processes, including retrotranslocation, the activation of membrane-anchored transcription factors, nuclear envelop formation, spindle disassembly, the homotypic fusion of the ER/Golgi membranes, DNA replication, and transcriptional regulation 96, 97, 98, 99, 100. The substrate specificity probably lies in the diverse p97 cofactors, each of which could in principle link p97 to a distinct set of substrates 97, 101.
The major cofactor that assists p97 in retrotranslocation is a dimer consisting of Ufd1 and Npl4, although there are known exceptions 102, 103. Different p97 cofactors may each affiliate with a specific type of retrotranslocation complex, allowing p97 to act on almost all ERAD substrates regardless of their translocation route. The p97-Ufd1-Npl4 complex contains several ubiquitin-binding motifs that each can recognize polyubiquitin signals on an ERAD substrate 104, 105, 106, 107. Moreover, p97 itself has an intrinsic affinity to unfolded substrates 108. Existing evidence suggests that p97/Cdc48 may first bind a non-modified, presumably unfolded substrate emerging from a translocation site. This interaction may serve as a ratcheting mechanism that prevents substrate from slipping back into the ER, and thus allows substrate to be efficiently ubiquitinated. Once ubiquitin chains are conjugated to the substrate, the ubiquitin signal is further recognized by the ubiquitin-binding motifs in the ATPase complex, leading to the extraction of polypeptides from the ER membrane 104.
Substrate delivery to the proteasome
It has been demonstrated that ERAD substrates are exported into the cytosol in an unfolded form, perhaps due to size limitation of the putative membrane retrotranslocons 36. Once released from the ER membrane, polypeptides need to be rapidly targeted to the proteasome for degradation. Otherwise, the dislocated ERAD products, many of which bear aggregation-prone hydrophobic transmembrane segments, may form toxic aggregates in the cytosol. It has been proposed that certain ubiquitin-binding factors may interact with the ATPase Cdc48/p97 and the proteasome in an alternate mode, leading to substrate hand-off from p97 to the proteasome 85. Such shuttling molecule candidates include Ufd2 and ataxin-3, which are remarkably distinct in biochemical activity: Ufd2 is an ubiquitin E4 enzyme that extends short polyubiquitin chains on a substrate 87, 109, whereas ataxin-3 is a member of the Joseph family of deubiquitinating enzymes that can trim or edit ubiquitin chains 110, 111, 112. Nevertheless, both molecules are able to interact with Cdc48/p97 as well as the UBA-UBL domain-bearing proteins Rad23 and Ubiquilin/Dsk2 113, 114. Rad23 and Ubiquilin/Dsk2 belong to a family of ubiquitin receptors that are docked on the proteasome to promote substrate-proteasome interactions. In yeast, it seems that several redundant mechanisms exist to channel ubiquitinated proteins to the proteasome. For example, Rad23p can cooperate with the N-glycanase Png1p to facilitate the degradation of glycoproteins and with Ufd2p to turnover non-glycoproteins 115. Little is known about how these factors shuttle substrates to the proteasome, but the fact that the delivery process can involve ubiquitin modifying enzymes with opposing activities underscores the importance of ubiquitin chain dynamics in this process.
The degradation machinery
The 26S proteasome is the major degradation machinery in the cell for dysfunctional or damaged proteins. Proteasomal degradation also regulates a variety of cellular process such as cell cycle progression. The proteasome is a large multi-subunit enzyme that is comprised of two sub-complexes, the 19S regulatory complex and the 20S proteolytic complex 116. The 20S sub-complex is made of four stacked rings in a α-β-β-α geometry with each ring containing either 7 α or β subunits. Together, they are assembled into a barrel-like structure that harbors a chymotrypsin-like, a trypsin-like, and a peptidyl-glutamyl peptide-hydrolyzing (PHGH) proteolytic activity inside the barrel. The 19S sub-complex is comprised of at least 19 proteins that can be further divided into two sub-assemblies. Among the 10 proteins in the base, six AAA ATPases are assembled into a hexameric ring that sits on top of the 20S sub-complex. These ATPases can unfold substrates to facilitate their entry into the degradation chamber. They also regulate the gated proteasome opening and provide the driving force that pulls substrates into the degradation chamber. The remaining proteins in the 19S particle form a lid on top of the base. The lid contains ubiquitin-binding sites that collect ubiquitinated proteins, interacts with substrate delivery factors, and harbors deubiquitinating activities that modulate ubiquitin conjugates on substrates en route to the destruction chamber.
Additional factors
Many additional factors have been identified to facilitate ERAD. Some of these factors are conserved from yeast to human, whereas others are specific for higher eukaryotes. In yeast, Usa1p was recently shown to function as a scaffold protein to recruit the Derlin ortholog Der1p protein to the ubiquitin ligase Hrd1p 117, 118. The mammalian homolog of Usa1p is Herp, which is also required for ERAD, but its precise function is unclear 38, 119. In yeast, the Cdc48p complex is recruited to the ER membrane via binding to the Ubx2p protein 120, 121, whereas in mammalian cells, several ER-associated membrane proteins contain high affinity binding sites for p97. These include the ubiquitin ligase gp78 122, a single-spanning membrane protein termed VIMP 53, the Ubx domain-containing protein UbxD8 123, and Erasin 124. These factors may act in parallel or sequentially to engage the p97 ATPase in ERAD. In mammals, several substrate-specific ERAD factors have also been identified. For example, a membrane protein complex comprised of SPFH1 and SPFH2 facilitates ubiquitination and degradation of inositol 1, 4, 5-trisphosphate receptors 125. A second example is Bap31, which promotes the degradation of a mutant variant of the cystic fibrosis transmembrane conductance regulator 126. The exact functions of these factors in ERAD are currently unclear, but their existence underscores the complexity, and perhaps the underlying specificity, of the ERAD system in higher eukaryotes.
ER stress: an intersection of life and death pathways
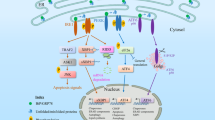
In addition to ERQC, eukaryotic cells have also evolved an integrated, adaptive stress response program, also known as the unfolded protein response (UPR) that helps alleviate protein-folding crises in the ER. This topic has been extensively reviewed 2, 127, 128, 129, 130. In brief, the core of the UPR is comprised of several signaling branches mediated by transmembrane proteins such as IRE1, PERK, and ATF6. These proteins sense protein misfolding in the ER lumen and then relay the signals to other parts of the cell to induce adaptive changes (see below and also Figure 2), which include transcriptional upregulation of genes involved in protein folding, degradation, and trafficking, transient inhibition of protein translation and translocation into the ER, decay of ER-localized mRNAs, and induction of autophagy 2, 131.

The two faces of the UPR signaling. In the mammalian ER, protein-misfolding stress is sensed by three membrane-associated proteins, ATF6, PERK, and IRE1. ATF6 is a transcription factor whose activation requires the translocation of ATF6 to the Golgi and the subsequent processing of ATF6 by two membrane proteases. PERK is a protein kinase that is autophosphorylated and activated when GRP78 dissociates from its ER luminal domain in response to ER stress. PERK phosphorylates eIF2α, leading to global attenuation of protein synthesis that only a few proteins including the transcription factor ATF4 can escape. ATF4 activates downstream genes that can have either a pro-survival or pro-apoptotic role (pro-death events are marked in blue whereas pro-survival events are marked in red). IRE1 is also a membrane bound protein kinase that is activated when GRP78 dissociates from it. Phosphorylation of IRE1 activates its endonuclease activity, which processes the XBP1 mRNA to produce a potent transcription factor. IRE1 endonuclease activity can also degrade ER-localized mRNA. The cytosolic domain of IRE1 can also bind TRAF2, which modulates the activity of two pro-apoptotic kinases, JNK and ASK1.
Although the unfolded protein response is elicited to promote cell viability under stress conditions, persistent ER stress can also switch on an apoptotic program to eliminate stressed cells. Intriguingly, the cyto-protective and the cyto-destructive signals are initiated by the same set of ER stress sensor proteins, raising the question of how a pro-survival or pro-death cell fate is determined under stress conditions.
The IRE1 signaling
Inositol-requiring enzyme 1 (IRE1), a Ser/Thr protein kinase and endoribonuclease, regulates the most evolutionarily conserved branch of the ER stress signaling network 132, 133. The ER luminal domain of IRE1 can bind BiP/GRP78, which prevents IRE1 oligomerization and maintains it in an inactive state 134, 135, 136. Upon ER stress induction, BiP dissociates from IRE1, allowing it to oligomerize 137. Moreover, IRE1 also contains a luminal MHC-like domain that may directly interact with misfolded proteins 130, 138. Recent studies suggest that at least in yeast direct interaction of Ire1p with misfolded proteins is essential for orienting the Ire1 oligomer into a proper configuration to activate its kinase and ribonuclease activities 139. In mammals, it appears that dissociation of BiP/GRP78 from IRE1 is sufficient to activate it 140. IRE1 activation results in the unconventional splicing of the mRNAs encoding the transcription factor XBP1 in mammals or Hac1p in yeast 141, 142, 143, 144, which results in the production of an activated form of the transcription factor and subsequent upregulation of genes involved in protein folding, degradation and trafficking. This improves both the ER folding capacity and ERAD efficiency of the cell. Lastly, recent studies reveal that IRE1 activation can activate autophagy 145, which presumably routes a fraction of misfolded ER proteins to the lysosome for degradation. These activities act together to restore ER proteostasis and thereby promote cell vitality under stress conditions.
In addition to its pro-survival actions, IRE1 signaling in mammalian cells also has a pro-apoptotic role. The cytosolic domain of IRE1 was reported to interact with the Ser/Thr kinase, tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2), which modulates the c-Jun N terminal kinase (JNK) activity to induce cell death 146. The IRE1-TRAF2 interaction also activates the apoptosis signal-regulating kinase 1 (Ask1) 147, which may contribute to the neuronal cell death induced by polyglutamine repeat-containing proteins. Moreover, a recent study shows that IRE1 autophosphorylation activates its RNAase activity, which not only catalyzes the splicing of XBP1 mRNA, but also induces the decay of many ER-localized mRNAs that encode secretory and membrane proteins 148, 150. Interestingly, the degradation of ER-localized mRNAs and XBP1 splicing are mechanistically uncoupled events that involve distinct modes of IRE1 RNAase actions. Accordingly, while overexpression of wild type IRE1, which induces both mRNA decay and XBP1 splicing, triggers apoptosis, an IRE1 mutant that only induces XBP1 splicing in the presence of a pseudokinase activator compound fails to induce cell death. The degradation of mRNAs encoding ER chaperones and other secretory proteins may be beneficial to cell survival at the initial phase of ER stress, as it helps reduce protein flux to the ER. However, it may become detrimental if persistent mRNA degradation leads to a depletion of ER chaperones, which would impair the ER folding capacity.
The PERK-eIF2α-ATF4 cascade
In addition to IRE1, the mammalian UPR also activates another ER-localized transmembrane protein kinase termed PERK [protein kinase RNA (PKR)-like ER kinase] 151. The ER luminal portion of PERK contains a stress-sensing domain that is both structurally and functionally related to that of IRE1 135. The cytoplasmic portion of PERK also has a protein kinase domain that is activated when PERK oligomerizes in stressed cells. Activated PERK phosphorylates the α-subunit of the eukaryotic translation initiation factor-2 (eIF2α), resulting in globally attenuated protein translation. This reduces the protein load the ER 152. In addition to translation inhibition, PERK also contributes to the adaptive stress response by influencing the gene expression landscape. A key player in this process is the cAMP response element-binding (CREB) transcription factor ATF4, which is selectively activated at the translational level despite the global translation inhibition in response to eIF2α phosphorylation. ATF4 upregulates the expression of many pro-survival genes including many ER chaperones, which helps cells adapt to the misfolding stress in the ER 152.
Interestingly, the ATF4 target genes also include some well-known pro-apoptotic factors such as CHOP/GADD153 and Noxa (see below) 153, 154, 155. CHOP/GADD153 is a transcription factor whose induction inhibits cell proliferation and induces apoptosis. CHOP can act as both a transcriptional repressor and activator. For example, it suppresses the expression of the multi-domain anti-apoptotic protein Bcl-2 156, but upregulates the expression of GADD34, a subunit of a phosphatase complex that dephosphorylates eIF2α 154. It was proposed that reduced eIF2α phosphorylation by the CHOP-mediated negative feedback loop may increase cell sensitivity to ER stress-induced apoptosis. In support of this model, GADD34-deficient cells have a sustained level of phosphorylated eIF2α and are more resistant to cytotoxicity imposed by ER stress 154. Moreover, a chemical inhibitor of the eIF2α phosphatase, called salubrinal, also protects cells against ER stress-induced apoptosis 157. CHOP also regulates the expression of the BH3-only pro-apoptotic protein Bim (see below) 158. Thus, a number of transcriptional changes are induced as a result of PERK activation that impact the cellular choice between survival and apoptosis.
The mechanism that translates the complex PERK-eIF2α-ATF4 signaling into either a pro-survival or a pro-death cellular action is unclear. It has been proposed that the duration of PERK signaling relative to that of XBP1 activation may influence this critical decision-making process. In response to prolonged ER stress, XBP1 splicing is attenuated, while the PERK activity is maintained 159. This may shift the cell towards a more destructive fate given the above mentioned connections between prolonged PERK signaling and the activation of pro-apoptotic proteins.
The Bcl-2 protein family
Mitochondria-initiated apoptosis is regulated by the Bcl-2 family proteins, which include Bax and Bak (two pro-apoptotic effectors that participate directly in the permeabilization of the mitochondrial outer membrane), anti-apoptotic factors such as Bcl-2, Bcl-xL, Mcl-1, and Bfl/A1, and factors termed BH3-only proteins. Many of the first two classes of Bcl-2 family proteins belong to the so-called tail-anchored proteins that contain a carboxyl-terminal transmembrane domain, which localizes these proteins to the mitochondrial outer membrane. In contrast, the BH3-only proteins do not have any transmembrane segment and contain only a Bcl-2 homology domain 3 (BH3). However, they can interact with both pro-apoptotic and pro-survival Bcl-2 family proteins to regulate their activities. Although some BH3-only proteins may promote apoptosis by antagonizing the function of the pro-survival Bcl-2 proteins, recent studies have suggested a direct role for at least some BH3-only proteins in the activation of Bax and Bak 160, 161, 162. Upon the binding of a BH3 helix to Bax, Bax undergoes a conformational change that mobilizes its carboxyl terminal helix for membrane translocation. Importantly, the BH3 domain of Bax is now exposed, which can interact with a yet-to-be activated Bax monomer to propagate the death signal within a Bax homo-oligomeric assembly 163.
Several Bcl-2 family proteins have established connections with the UPR signaling at the ER. It is apparent that many of these BH3 proteins act as downstream executors to initiate cell death in response to an overwhelming ER stress signal. In addition, direct interactions between some Bcl-2 family proteins and ER stress signaling molecules were observed. Specifically, the pro-apoptotic Bcl-2 family proteins Bax and Bak were found to be physically present at the ER membrane in addition to their mitochondrial localization. These ER-associated Bcl-2 family proteins were reported to interact with IRE1 to modulate its signaling properties upon ER stress induction. Accordingly, in Bax and Bak double knock-out mice, the IRE1 branch of the UPR signaling was impaired 164. Along the same line, an ER-localized natural Bak inhibitory polypeptide (BI-1) also forms a complex with IRE1, and this interaction suppresses IRE1 signaling during ER stress 165. The ER pool of Bax and Bak may also regulate an ER-to-mitochondria calcium flux in response to stress signals, which may also contribute to apoptosis 166, 167.
The expression of many BH3-only proteins including Bim, Puma and Noxa, is induced by ER stress. The mechanisms by which UPR regulates the expression of these genes are variable. Even for a given BH3-only protein, its induction can be mediated by different mechanisms in different cell types or in response to different stress stimuli. For example, Noxa upregulation in mouse embryonic fibroblasts (MEFs) or human melanoma cells treated with the ER stressors thapsigargin or tunicamycin appears to be dependent on the tumor suppressor p53 168, 169. However, in neuroblastoma and melanoma cells treated with the chemotherapeutic agents feretinide or in transformed 293T cells exposed to the ERAD inhibitors Eeyarestatin I (EerI) and bortezomib, Noxa induction is independent of p53 155, 170, 171, 172. Instead, Noxa up-regulation involves the ER stress-regulated transcription factors ATF4 and ATF3 155, 170 (see below). In addition, the oncogene c-Myc was shown to be required for bortezomib-induced Noxa expression 172. Likewise, both p53-dependent and -independent mechanisms have been reported for the induction of PUMA during ER stress 168, 169, 173. For Bim, it has been shown that ER stress elevates Bim protein levels by two independent mechanisms. ER stress can reduce proteasomal degradation of Bim, while at the same time the UPR signal can activate CHOP, which in turn upregulates Bim mRNA expression 158. Clearly, components of the mitochondria-initiated apoptotic pathway are closely integrated with the UPR program.
Targeting ER proteostasis in cancer therapy
Cells bearing different secretory capacities and basal levels of ER stress can differ significantly in sensitivity to ER stress-induced cell death. Certain cancers such as multiple myeloma are particularly sensitive to ER stress-induced cell death, perhaps because these cells constantly carry a high secretory load due to their specialized role in antibody production. Thus, a therapeutic window exists that allows some ER stress inducers to selectively kill these malignant cells without imposing significant damage to surrounding healthy cells. In this section, we discuss the potential cancer treatment strategies that target ER proteostasis.
The proteasome
Bortezomib (VelcadeTM, also named PS-341) is a first-in-class proteasome inhibitor that acts at least in part by targeting ER proteostasis to treat cancer. Bortezomib is a peptide boronic acid analog initially designed to inhibit the chymotrypsin activity of the proteasome by mimicking substrate binding. It was shown to be a potent inhibitor of the pro-inflammatory NFκB signaling pathway due to inhibition of IκB degradation 174. Subsequent studies in the National Cancer Institute (NCI) set of 60 cancer cell lines demonstrated broad spectrum anti-tumor activities for bortezomib 175, which were later confirmed in a mouse xenograft model 176. Following rigorous clinical testing, the United States Food and Drug Administration (FDA) approved the use of bortezomib for the treatment of multiple myeloma in 2003 and later extended its use to mantle cell lymphoma.
The approval of bortezomib by the FDA as an anti-cancer agent has fueled the interest in understanding the mechanism underlying its anti-cancer activity. Given the diverse cellular activities of proteasomal substrates, it is not surprising to learn that many cellular pathways contribute to bortezomib-induced cell death. Moreover, the cellular factors involved in bortezomib-induced cell death can vary among different cell types. For example, bortezomib acts through p53 to induce growth arrest and cell death in mammary epithelial cells 177, but in PC-3 prostate cancer cells, the p53 function becomes dispensable for bortezomib-induced cytotoxicity 178. It was initially thought that inhibition of the pro-survival NFκB signaling pathway might be the predominant cause of bortezomib-induced cell death. However, subsequent studies comparing the activities of bortezomib with an NFκB-specific inhibitor showed that NFκB inhibition by itself cannot fully account for the anti-tumor activity of bortezomib 179.
Recent studies have underscored the importance of the pro-apoptotic protein Noxa in bortezomib-mediated cytotoxicity. In bortezomib-treated cells, Noxa mRNA and protein levels are dramatically increased. Knock down of Noxa in a variety of transformed cell lines renders resistance to bortezomib-induced apoptosis 172, 180, 181, 182, 183, 184. Although Noxa was initially discovered as a transcriptional target of p53 185, the mechanism that activates Noxa in bortezomib-treated cells appears to be independent of p53. Instead, the UPR plays a critical role. Specifically, bortezomib activates the PERK branch of the UPR, leading to an upregulation of the transcription factors ATF4 and ATF3, which form a hetero-oligomer on the Noxa promoter. In addition, bortezomib attenuates ubiquitination of histone H2A to relieve its inhibitory effect on Noxa transcription 155. These observations establish an important link between ER stress and the anti-cancer action of bortezomib. Consistent with this idea, several recent studies show that the UPR induction upon proteasome inhibition is essential for bortezomib-induced cytotoxicity 186, 187, 188, 189. Together, these studies highlight an important role for the UPR pathway in the anti-cancer action of bortezomib, and suggest that ER proteostasis can be targeted in anti-cancer therapies.
p97 and associated deubiquitinating enzymes
As discussed above, the ERQC program employs several independent mechanisms for substrate recognition and retrotranslocation from the ER. However, the retrotranslocation of almost all ERAD substrates converges on the p97 ATPase for membrane extraction and for subsequent transfer to the proteasome 90, 97. Accordingly, inhibition of p97 and the proteasome usually generates a more pronounced effect on ER homeostasis than interference with an upstream ERAD step. Therefore, it is conceivable that p97 may be a potential target for cancer therapy. In support of this hypothesis, recent studies have demonstrated that an ERAD-specific inhibitor, termed Eeyarestatin I (EerI), which targets p97, can induce cell death in hematologic cancer cells via a mechanism similar to that of bortezomib 155, 190. Like bortezomib, EerI induces ER stress and causes downregulation of histone H2A ubiquitination, which result in Noxa activation and cell death. The anti-tumor profile of EerI in the NCI 60 cancer cell lines is similar to that of bortezomib (Figure 3) 175. Importantly, EerI can dramatically synergize with bortezomib to induce cancer cell death 155. These results show that inhibition of p97 can achieve a similar anti-cancer effect as blocking the proteasome. Indeed, interest in searching for more potent inhibitors that block p97 ATPase activity is mounting 191.

The p97 inhibitor EerI has broad spectrum anti-cancer activities. The NCI 60 cancer cells were treated with EerI at 5 doses ranging from 10 nM to 100 μM. Concentration-response curves from two independent experiments were used to calculate the average GI50 (the concentration of drug required to obtain 50% of growth inhibition) for each cell line. The graph shows the GI50 of each cell line relative to the mean value.
EerI is a bi-modular compound that is comprised of two functionally independent domains. An aromatic module targets EerI to the ER membrane, allowing a nitrofuran-containing (NFC) module to directly bind to p97 and to interfere with its membrane-associated functions. As a result, EerI is a much more specific disruptor of ER proteostasis compared to a compound that only has the NFC domain 192. EerI does not block the nucleotide hydrolysis cycle of p97. Instead, the binding of EerI to p97 induces a conformational change in p97, which may alter its interactions with cofactors 192. The phenotypic consequence of EerI binding to p97 is complex. In cells exposed to EerI for a prolonged period, p97-mediated retrotranslocation is completely blocked 193. However, in the initial phase of EerI treatment, a major consequence is the disruption of ubiquitin homeostasis as polyubiquitinated proteins accumulate. This phenotype may be due to an indirect effect of EerI on the action of some deubiquitinating enzymes bound to p97 190. These deubiquitinating enzymes can act either upstream (e.g., YOD1) 194 or downstream (e.g., ataxin-3) of the p97 ATPase cycle to modulate the ubiquitin contents on retrotranslocation substrates 110. EerI's capability to influence deubiquitinating activities associated with p97 is critical for its cytotoxic action, and disruption of ubiquitin homeostasis due to insufficient deubiquitination may be the cause for the loss of ubiquitinated histone H2A. In this regard, chemical inhibitors directly targeting p97-associated deubiquitinating enzymes may achieve similar anti-cancer effects as EerI.
Other anti-cancer targets
Given the essential role of the IRE1 endonuclease activity in promoting cell vitality under ER stress conditions, it has been thought that inhibitors targeting this nuclease activity could have anti-cancer activities 195. Indeed, Feldman and Koong recently reported the identification of the first-in-class IRE1 endonuclease inhibitors, which they named Irestatins. These compounds were shown to be potent cell death inducers, particularly for oxygen-starved cancer cells. One of these compounds was also shown to have anti-cancer activity in a mouse xenograft model 196. Another potential anti-cancer target in the UPR signaling network is the PERK kinase 197 given the precedent success in developing kinase inhibitors for cancer therapy. However, no potent inhibitor for this enzyme has been reported so far despite some serious efforts from several research groups.
Perspectives
Researchers over the past decade have made tremendous progress towards a better understanding of how eukaryotic cells cope with protein misfolding in the ER and how deregulation of ER proteostasis can cause cell dysfunction and death. The knowledge obtained to date has, no doubt, fueled the development of novel cancer therapeutics concepts, which are now being transformed into potential new medicines. Nonetheless, many fundamental aspects regarding ER proteostasis regulation remain poorly understood. For example, it is still unclear how the cell distinguishes misfolded ER proteins from those in the midst of the folding process. How misfolded proteins are moved across the ER membrane during retrotranslocation is also unknown. In addition, we are almost completely ignorant about the regulation of ERAD capacity in various cell types or in cells facing different stress challenges. In particular, understanding the precise mechanism that governs the life or death decision-making process of stressed cells, such as those stressed by bortezomib, may provide a better guide to developing new treatments that are more effective in battling against cancer.
References
Vembar SS, Brodsky JL . One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 2008; 9:944–957.
Ron D, Walter P . Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519–529.
Rutkowski DT, Kaufman RJ . A trip to the ER: coping with stress. Trends Cell Biol 2004; 14:20–28.
Boyce M, Yuan J . Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ 2006; 13:363–373.
Perez-Galan P, Mora-Jensen H, Weniger MA, et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood 2011; 117:542–552.
Koumenis C . ER stress, hypoxia tolerance and tumor progression. Curr Mol Med 2006; 6:55–69.
Boelens J, Lust S, Offner F, Bracke ME, Vanhoecke BW . Review. The endoplasmic reticulum: a target for new anticancer drugs. In Vivo 2007; 21:215–226.
Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T . The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009; 458:453–460.
Ye Y . The role of the ubiquitin-proteasome system in ER quality control. Essays Biochem 2005; 41:99–112.
Bukau B, Weissman J, Horwich A . Molecular chaperones and protein quality control. Cell 2006; 125:443–451.
Nakatsukasa K, Brodsky JL . The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 2008; 9:861–870.
Vashist S, Ng DT . Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 2004; 165:41–52.
Taxis C, Hitt R, Park SH, Deak PM, Kostova Z, Wolf DH . Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem 2003; 278:35903–35913.
Carvalho P, Goder V, Rapoport TA . Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006; 126:361–373.
Swanson R, Locher M, Hochstrasser M . A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev 2001; 15:2660–2674.
Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M . Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol 2010; 188:223–235.
Maattanen P, Kozlov G, Gehring K, Thomas DY . ERp57 and PDI: multifunctional protein disulfide isomerases with similar domain architectures but differing substrate-partner associations. Biochem Cell Biol 2006; 84:881–889.
Hammond C, Helenius A . Folding of VSV G protein: sequential interaction with BiP and calnexin. Science 1994; 266:456–458.
Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P . Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol 2002; 158:247–257.
Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B . Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J 1997; 16:1501–1507.
Erbse A, Mayer MP, Bukau B . Mechanism of substrate recognition by Hsp70 chaperones. Biochem Soc Trans 2004; 32:617–621.
Matlack KE, Misselwitz B, Plath K, Rapoport TA . BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell 1999; 97:553–564.
Parodi AJ . Protein glucosylation and its role in protein folding. Annu Rev Biochem 2000; 69:69–93.
Lederkremer GZ . Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol 2009; 19:515–523.
Hosokawa N, Wada I, Hasegawa K, et al. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep 2001; 2:415–422.
Oda Y, Hosokawa N, Wada I, Nagata K . EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 2003; 299:1394–1397.
Molinari M, Calanca V, Galli C, Lucca P, Paganetti P . Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003; 299:1397–1400.
Quan EM, Kamiya Y, Kamiya D, et al. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell 2008; 32:870–877.
Clerc S, Hirsch C, Oggier DM, et al. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol 2009; 184:159–172.
Mikami K, Yamaguchi D, Tateno H, et al. The sugar-binding ability of human OS-9 and its involvement in ER-associated degradation. Glycobiology 2010; 20:310–321.
Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA . Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell 2005; 19:765–775.
Kim W, Spear ED, Ng DT . Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell 2005; 19:753–764.
Gauss R, Jarosch E, Sommer T, Hirsch C . A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol 2006; 8:849–854.
Kanehara K, Kawaguchi S, Ng DT . The EDEM and Yos9p families of lectin-like ERAD factors. Semin Cell Dev Biol 2007; 6:743–750.
Spear ED, Ng DT . Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J Cell Biol 2005; 169:73–82.
Bhamidipati A, Denic V, Quan EM, Weissman JS . Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell 2005; 19:741–751.
Soetandyo N, Wang Q, Ye Y, Li L . Role of intramembrane charged residues in the quality control of unassembled T-cell receptor alpha-chains at the endoplasmic reticulum. J Cell Sci 2010; 123:1031–1038.
Okuda-Shimizu Y, Hendershot LM . Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell 2007; 28:544–554.
Lee SO, Cho K, Cho S, Kim I, Oh C, Ahn K . Protein disulphide isomerase is required for signal peptide peptidase-mediated protein degradation. EMBO J 2010; 29:363–375.
Bernardi KM, Forster ML, Lencer WI, Tsai B . Derlin-1 facilitates the retro-translocation of cholera toxin. Mol Biol Cell 2008; 19:877–884.
Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K . ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008; 321:569–572.
Ploegh HL . A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 2007; 448:435–438.
Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH . Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 1997; 388:891–895.
Zhou M, Schekman R . The engagement of Sec61p in the ER dislocation process. Mol Cell 1999; 4:925–934.
Scott DC, Schekman R . Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol 2008; 181:1095–1105.
Wahlman J, DeMartino GN, Skach WR, Bulleid NJ, Brodsky JL, Johnson AE . Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell 2007; 129:943–955.
Huyer G, Piluek WF, Fansler Z, et al. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem 2004; 279:38369–38378.
Walter J, Urban J, Volkwein C, Sommer T . Sec61p-independent degradation of the tail-anchored ER membrane protein Ubc6p. EMBO J 2001; 20:3124–3131.
Garza RM, Sato BK, Hampton RY . In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem 2009; 284:14710–14722.
Lilley BN, Ploegh HL . Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 2005; 102:14296–14301.
Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA . Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 2005; 102:14132–14138.
Lilley BN, Ploegh HL . A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004; 429:834–840.
Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA . A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004; 429:841–847.
Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K . Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol 2006; 172:383–393.
Katiyar S, Joshi S, Lennarz WJ . The retrotranslocation protein Derlin-1 binds peptide:N-glycanase to the endoplasmic reticulum. Mol Biol Cell 2005; 16:4584–4594.
Gauss R, Sommer T, Jarosch E . The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J 2006; 25:1827–1835.
Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY . Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 2001; 3:24–29.
Sato BK, Schulz D, Do PH, Hampton RY . Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell 2009; 34:212–222.
Carvalho P, Stanley AM, Rapoport TA . Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010; 143:579–591.
Stagg HR, Thomas M, van den Boomen D, et al. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol 2009; 186:685–692.
Loureiro J, Lilley BN, Spooner E, Noriega V, Tortorella D, Ploegh HL . Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature 2006; 441:894–897.
Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJ, Hansen TH . Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol 2007; 177:613–624.
Li W, Ye Y . Polyubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci 2008; 65:2397–2406.
Hiller MM, Finger A, Schweiger M, Wolf DH . ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 1996; 273:1725–1728.
Biederer T, Volkwein C, Sommer T . Role of Cue1p in ubiquitination and degradation at the ER surface. Science 1997; 278:1806–1809.
Shamu CE, Flierman D, Ploegh HL, Rapoport TA, Chau V . Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol Biol Cell 2001; 12:2546–2555.
Sommer T, Jentsch S . A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 1993; 365:176–179.
Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T . A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol 2000; 2:379–384.
Ye Y, Rape M . Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol 2009; 10:755–764.
Tiwari S, Weissman AM . Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J Biol Chem 2001; 276:16193–16200.
Lenk U, Yu H, Walter J, et al. A role for mammalian Ubc6 homologues in ER-associated protein degradation. J Cell Sci 2002; 115:3007–3014.
Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM . The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci USA 2006; 103:341–346.
Tsai YC, Mendoza A, Mariano JM, et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med 2007; 13:1504–1509.
Wang X, Herr RA, Rabelink M, Hoeben RC, Wiertz EJ, Hansen TH . Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J Cell Biol 2009; 187:655–668.
Kikkert M, Doolman R, Dai M, et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem 2004; 279:3525–3534.
Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM . The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA 2001; 98:14422–14427.
Shen Y, Ballar P, Fang S . Ubiquitin ligase gp78 increases solubility and facilitates degradation of the Z variant of alpha-1-antitrypsin. Biochem Biophys Res Commun 2006; 349:1285–1293.
Song BL, Sever N, DeBose-Boyd RA . Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell 2005; 19:829–840.
Morito D, Hirao K, Oda Y, et al. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell 2008; 19:1328–1336.
Younger JM, Chen L, Ren HY, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006; 126:571–582.
Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM . The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 2001; 3:100–105.
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R . An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001; 105:891–902.
Yoshida Y, Chiba T, Tokunaga F, et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002; 418:438–442.
Yoshida Y, Tokunaga F, Chiba T, Iwai K, Tanaka K, Tai T . Fbs2 is a new member of the E3 ubiquitin ligase family that recognizes sugar chains. J Biol Chem 2003; 278:43877–43884.
Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S . A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 2005; 120:73–84.
Tu D, Li W, Ye Y, Brunger AT . Inaugural Article: Structure and function of the yeast U-box-containing ubiquitin ligase Ufd2p. Proc Natl Acad Sci USA 2007; 104:15599–15606.
Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S . A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 1999; 96:635–644.
Li W, Tu D, Li L, et al. Mechanistic insights into active site-associated polyubiquitination by the ubiquitin-conjugating enzyme Ube2g2. Proc Natl Acad Sci USA 2009; 106:3722–3727.
Das R, Mariano J, Tsai YC, et al. Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol Cell 2009; 34:674–685.
Ye Y, Meyer HH, Rapoport TA . The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001; 414:652–656.
Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY . HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell 2001; 12:4114–4128.
Jarosch E, Geiss-Friedlander R, Meusser B, Walter J, Sommer T . Protein dislocation from the endoplasmic reticulum--pulling out the suspect. Traffic 2002; 3:530–536.
Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S . AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 2002; 22:626–634.
DeLaBarre B, Brunger AT . Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol 2003; 10:856–863.
Zhang X, Shaw A, Bates PA, et al. Structure of the AAA ATPase p97. Mol Cell 2000; 6:1473–1484.
Jentsch S, Rumpf S . Cdc48 (p97): a "molecular gearbox" in the ubiquitin pathway? Trends Biochem Sci 2007; 32:6–11.
Ye Y . Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J Struct Biol 2006; 156:29–40.
Ramadan K, Bruderer R, Spiga FM, et al. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 2007; 450:1258–1262.
Mouysset J, Deichsel A, Moser S, et al. Cell cycle progression requires the CDC-48UFD-1/NPL-4 complex for efficient DNA replication. Proc Natl Acad Sci USA 2008; 105:12879–12884.
Wilcox AJ, Laney JD . A ubiquitin-selective AAA-ATPase mediates transcriptional switching by remodelling a repressor-promoter DNA complex. Nat Cell Biol 2009; 11:1481–1486.
Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G . A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 2000; 19:2181–2192.
Soetandyo N, Ye Y . The p97 ATPase dislocates MHC class I heavy chain in US2 expressing cells via an Ufd1-Npl4 independent mechanism. J Biol Chem 2010; 285:32352–32359.
Ballar P, Shen Y, Yang H, Fang S . The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J Biol Chem 2006; 281:35359–35368.
Ye Y, Meyer HH, Rapoport TA . Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 2003; 162:71–84.
Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S . Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 2001; 107:667–677.
Meyer HH, Wang Y, Warren G . Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J 2002; 21:5645–5652.
Dai RM, Li CC . Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 2001; 3:740–744.
Thoms S . Cdc48 can distinguish between native and non-native proteins in the absence of cofactors. FEBS Lett 2002; 520:107–110.
Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL . Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 2008; 132:101–112.
Wang Q, Li L, Ye Y . Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol 2006; 174:963–971.
Zhong X, Pittman RN . Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum Mol Genet 2006; 15:2409–2420.
Winborn BJ, Travis SM, Todi SV, et al. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem 2008; 283:26436–26443.
Doss-Pepe EW, Stenroos ES, Johnson WG, Madura K . Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol Cell Biol 2003; 23:6469–6483.
Hanzelmann P, Stingele J, Hofmann K, Schindelin H, Raasi S . The yeast E4 ubiquitin ligase Ufd2 interacts with the ubiquitin-like domains of Rad23 and Dsk2 via a novel and distinct ubiquitin-like binding domain. J Biol Chem 2010; 285:20390–20398.
Kim I, Ahn J, Liu C, et al. The Png1-Rad23 complex regulates glycoprotein turnover. J Cell Biol 2006; 172:211–219.
Pickart CM, Cohen RE . Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 2004; 5:177–187.
Knop M, Finger A, Braun T, Hellmuth K, Wolf DH . Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J 1996; 15:753–763.
Horn SC, Hanna J, Hirsch C, et al. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell 2009; 36:782–793.
Schulze A, Standera S, Buerger E, et al. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol 2005; 354:1021–1027.
Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T . Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol 2005; 7:993–998.
Schuberth C, Buchberger A . Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol 2005; 7:999–1006.
Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S . AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 2004; 279:45676–45684.
Lee JN, Zhang X, Feramisco JD, Gong Y, Ye J . Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J Biol Chem 2008; 283:33772–33783.
Lim PJ, Danner R, Liang J, et al. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol 2009; 187:201–217.
Wang Y, Pearce MM, Sliter DA, et al. SPFH1 and SPFH2 mediate the ubiquitination and degradation of inositol 1,4,5-trisphosphate receptors in muscarinic receptor-expressing HeLa cells. Biochim Biophys Acta 2009; 1793:1710–1718.
Wang B, Heath-Engel H, Zhang D, et al. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 2008; 133:1080–1092.
Schroder M, Kaufman RJ . The mammalian unfolded protein response. Annu Rev Biochem 2005; 74:739–789.
Kaufman RJ . Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999; 13:1211–1233.
Schroder M, Kaufman RJ . Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr Mol Med 2006; 6:5–36.
Hetz C, Glimcher LH . Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell 2009; 35:551–561.
Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS . Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell 2006; 127:999–1013.
Cox JS, Shamu CE, Walter P . Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993; 73:1197–1206.
Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D . Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J 1998; 17:5708–5717.
Pincus D, Chevalier MW, Aragon T, et al. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol 2010; 8:e1000415.
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D . Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2:326–332.
Kimata Y, Kimata YI, Shimizu Y, et al. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell 2003; 14:2559–2569.
Korennykh AV, Egea PF, Korostelev AA, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009; 457:687–693.
Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P . On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA 2005; 102:18773–18784.
Kimata Y, Ishiwata-Kimata Y, Ito T, et al. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol 2007; 179:75–86.
Oikawa D, Kimata Y, Kohno K, Iwawaki T . Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res 2009; 315:2496–2504.
Sidrauski C, Walter P . The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997; 90:1031–1039.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K . XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107:881–891.
Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002; 415:92–96.
Shen X, Ellis RE, Lee K, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001; 107:893–903.
Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220–9231.
Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000; 287:664–666.
Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 2002; 16:1345–1355.
Hollien J, Weissman JS . Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006; 313:104–107.
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS . Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 2009; 186:323–331.
Han D, Lerner AG, Vande Walle L, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009; 138:562–575.
Harding HP, Zhang Y, Ron D . Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397:271–274.
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D . Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 2000; 5:897–904.
Ma Y, Brewer JW, Diehl JA, Hendershot LM . Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 2002; 318:1351–1365.
Novoa I, Zeng H, Harding HP, Ron D . Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 2001; 153:1011–1022.
Wang Q, Mora-Jensen H, Weniger MA, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci USA 2009; 106:2200–2205.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ . Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21:1249–1259.
Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005; 307:935–939.
Puthalakath H, O'Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007; 129:1337–1349.
Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318:944–949.
Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB . BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 2001; 15:1481–1486.
Tait SW, Green DR . Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11:621–632.
Kim H, Tu HC, Ren D, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 2009; 36:487–499.
Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD . BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell 2010; 40:481–492.
Hetz C, Bernasconi P, Fisher J, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006; 312:572–576.
Lisbona F, Rojas-Rivera D, Thielen P, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 2009; 33:679–691.
Scorrano L, Oakes SA, Opferman JT, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300:135–139.
Klee M, Pallauf K, Alcala S, Fleischer A, Pimentel-Muinos FX . Mitochondrial apoptosis induced by BH3-only molecules in the exclusive presence of endoplasmic reticular Bak. EMBO J 2009; 28:1757–1768.
Li J, Lee B, Lee AS . Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 2006; 281:7260–7270.
Jiang CC, Lucas K, Avery-Kiejda KA, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res 2008; 68:6708–6717.
Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP . Regulation of ER stress-induced cell death by ATF4 in neuroectodermal tumour cells. J Biol Chem 2009; 285:6091–6100.
Armstrong JL, Veal GJ, Redfern CP, Lovat PE . Role of Noxa in p53-independent fenretinide-induced apoptosis of neuroectodermal tumours. Apoptosis 2007; 12:613–622.
Nikiforov MA, Riblett M, Tang WH, et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc Natl Acad Sci USA 2007; 104:19488–19493.
Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH . Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol 2003; 162:587–597.
Palombella VJ, Conner EM, Fuseler JW, et al. Role of the proteasome and NF-kappaB in streptococcal cell wall-induced polyarthritis. Proc Natl Acad Sci USA 1998; 95:15671–15676.
Shoemaker RH . The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 2006; 6:813–823.
LeBlanc R, Catley LP, Hideshima T, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res 2002; 62:4996–5000.
MacLaren AP, Chapman RS, Wyllie AH, Watson CJ . p53-dependent apoptosis induced by proteasome inhibition in mammary epithelial cells. Cell Death Differ 2001; 8:210–218.
Adams J, Palombella VJ, Sausville EA, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999; 59:2615–2622.
Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem 2002; 277:16639–16647.
Fernandez Y, Verhaegen M, Miller TP, et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res 2005; 65:6294–6304.
Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res 2005; 65:6282–6293.
Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D . The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 2006; 107:257–264.
Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, et al. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res 2007; 67:5418–5424.
Rizzatti EG, Mora-Jensen H, Weniger MA, et al. Noxa mediates bortezomib induced apoptosis in both sensitive and intrinsically resistant mantle cell lymphoma cells and this effect is independent of constitutive activity of the AKT and NF-kappaB pathways. Leuk Lymphoma 2008; 49:798–808.
Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288:1053–1058.
Fribley AM, Evenchik B, Zeng Q, et al. Proteasome inhibitor PS-341 induces apoptosis in cisplatin-resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem 2006; 281:31440–31447.
Fribley A, Wang CY . Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol Ther 2006; 5:745–748.
Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr., Lee KP, Boise LH . Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107:4907–4916.
Fels DR, Ye J, Segan AT, et al. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Res 2008; 68:9323–9330.
Wang Q, Li L, Ye Y . Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem 2008; 283:7445–7554
Bursavich MG, Parker DP, Willardsen JA, et al. 2-Anilino-4-aryl-1,3-thiazole inhibitors of valosin-containing protein (VCP or p97). Bioorg Med Chem Lett 2010; 20:1677–1679.
Wang Q, Shinkre BA, Lee JG, et al. The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PLoS One 2010; 5:e15479.
Fiebiger E, Hirsch C, Vyas JM, Gordon E, Ploegh HL, Tortorella D . Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell 2004; 15:1635–1646.
Ernst R, Mueller B, Ploegh HL, Schlieker C . The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell 2009; 36:28–38.
Koong AC, Chauhan V, Romero-Ramirez L . Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol Ther 2006; 5:756–759.
Feldman DE, Koong AC . Irestatin, a potent inhibitor of IRE1{alpha} and the unfolded protein response, is a hypoxia-selective cytotoxin and impairs tumor growth. Journal of Clinical Oncology 2007; 25:3514.
Fels DR, Koumenis C . The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 2006; 5:723–728.
Acknowledgements
The authors' research is supported by the NIH intramural AIDS Targeted Antiviral Program (IATAP) and by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). We thank Michael Krause (NIDDK) for critical reading of the manuscript. We also thank the Developmental Therapeutics Program (DTP)/Division of Cancer Treatment and Diagnosis (DCTD) at National Cancer Institute (NCI) (http://dtp.cancer.gov) for testing compounds in the NCI 60 cancer cell lines. We apologize for not being able to cite all the relevant publications due to space limits.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Y., Ye, Y. Proteostasis regulation at the endoplasmic reticulum: a new perturbation site for targeted cancer therapy. Cell Res 21, 867–883 (2011). https://doi.org/10.1038/cr.2011.75
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.75
Keywords
This article is cited by
-
Integrating network pharmacology and experimental models to examine the mechanisms of corosolic acid in preventing hepatocellular carcinoma progression through activation PERK-eIF2a-ATF4 signaling
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Attenuation of Activated eIF2α Signaling by ISRIB Treatment After Spinal Cord Injury Improves Locomotor Function
Journal of Molecular Neuroscience (2022)
-
TRIM59 guards ER proteostasis and prevents Bortezomib-mediated colorectal cancer (CRC) cells’ killing
Investigational New Drugs (2022)
-
ARVib suppresses growth of advanced prostate cancer via inhibition of androgen receptor signaling
Oncogene (2021)
-
Investigating Chaperonin-Containing TCP-1 subunit 2 as an essential component of the chaperonin complex for tumorigenesis
Scientific Reports (2020)
