
Abstract
Plasma membrane (PM) blebs are dynamic actin-rich cell protrusions that occur, e.g., during cytokinesis, amoeboid cell motility and cell attachment. Using a targeted siRNA screen against 21 actin nucleation factors, we identify a novel and essential role of the human diaphanous formin DIAPH3 in PM blebbing during cell adhesion. Suppression of DIAPH3 inhibited blebbing to promote rapid cell spreading involving β1-integrin. Multiple isoforms of DIAPH3 were detected on the mRNA and protein level of which isoforms 3 and 7 were the largest and most abundant isoforms that however did not induce formation of actin-rich protrusions. Rather, PM blebbing specifically involved the low abundance isoform 1 of DIAPH3 and activation of isoform 7 by deletion of the diaphanous-autoregulatory domain caused the formation of filopodia. Dimerization and actin assembly activity were essential for induction of specific cell protrusions by DIAPH3 isoforms 1 and 7. Our data suggest that the N-terminal region comprising the GTPase-binding domain determined the subcellular localization of the formin as well as its protrusion activity between blebs and filopodia. We propose that isoform-selective actin assembly by DIAPH3 exerts specific and differentially regulated functions during cell adhesion and motility.
Similar content being viewed by others


Introduction
In response to extracellular cues, the plasma membrane (PM) of mammalian cells is subject to dynamic remodeling resulting in the formation of a diverse set of cell protrusions including lamellipodia, podosomes, filopodia and blebs 1, 2, 3, 4. Despite their distinct morphologies, formation of all these cell protrusions is governed by dynamic F-actin remodeling including de novo formation of actin filaments. Generation of new actin filaments is mediated by a diverse set of cellular machines referred to as actin nucleators. In mammalian cells, more than 20 different proteins have been identified thus far that bear the intrinsic ability to mediate nucleation of actin filaments, and that based on their domain organization and nucleation strategy, can be categorized into distinct groups 5. Among these, Arp2/3 represents an actin nucleation machinery acting as a protein assembly composed of seven subunits that caps pointed ends of actin filaments to promote filament elongation at the barbed end 6. Arp2/3 activity is stimulated by nucleation-promoting factors, which in the case of JMY can bear actin nucleation activity themselves 7. As an independent actin nucleator, JMY is part of a group of nucleators that recruit G-actin monomers via tandem repeats of WH2 domains, and the group includes the Spire, cordon-blue and the muscle-specific leiomodin families, the precise actin nucleation mechanism of which is still a matter of debate 8. Finally, the formin group of nucleators mediates actin polymerization by their signature formin homology (FH)2 domain, which, facilitated by the recruitment of profilin-actin by the adjacent FH1 domain, elongates F-actin filaments from the barbed end via a stair-step mechanism that critically depends on the dimerization of the FH2 domain 9, 10. In addition to these domains, that mediate actin polymerization, the so-called diaphanous formins are subject to autoinhibition 11 mediated by interaction of their C-terminal diaphanous autoregulatory domain (DAD) with an N-terminal region also referred to as diaphanous inhibitory domain (DID) that is part of the FH3 module. Activation through release of autoinhibition can occur via phosphorylation within the DAD, as demonstrated for FHOD1 12, 13, or by interaction of the N-terminal GTPase-binding domain (GBD) with GTPases of the Rho family as exemplified for mDia1 11.
While the dynamics and molecular regulation of lamellipodia and filopodia have been extensively studied, still relatively little is known about actin assembly factors in the formation of non-apoptoptic PM blebs in human cells. The term PM bleb refers to bulky protrusions of the PM that have been observed as early as 1919 (14, reviewed in 4). Subsequently, studies on blebs mainly focused on apoptotic cells that display large bleb-like protrusions as byproduct of the cell death process 15, 16. More recently, research has re-focused on non-apoptotic blebs that are characterized by their smaller size and more uniform morphology than those of apoptotic cells. Such PM blebs are observed during physiologically distinct processes such as cytokinesis, amoeboid types of invasive motility, and cell adhesion 17, 18, 19. Blebbing can also be induced experimentally, e.g. by disruption of the genes encoding the actin-membrane crosslinker filamin A or the tumor suppressor p53, or by overexpression of SH4 domain membrane targeting signals 12, 20, 21, 22, 23, 24, 25. Despite their distinct physiological context and triggers, PM blebs typically display high dynamics of bleb expansion and retraction. Bleb halt and retraction rely on acto-myosin contractility generated by Rho-ROCK signaling. Hence, rapid actin assembly as well as Rho/ROCK activity are essential for most, if not all, types of blebbing analyzed thus far 4, 26. The identity of specific actin nucleators required for distinct modes of blebbing, however, has remained a key open question.
Since all currently identified players involved in PM blebbing also represent core components for the formation of different types of cell protrusions, it remains unclear how such a wide plasticity of distinct cell protrusions can be generated by a common machinery. Furthermore, the lack of identification of molecular components with exclusive and specific roles in blebbing represents a major limitation to analyses of the physiological roles of PM blebbing. Conceivably, the involvement of a distinct and protrusion-specific combination of actin nucleators may constitute a versatile means for such diversification. As a largely unexplored source for generation of even further diversity, multiple isoforms exist for many actin nucleators. Using a targeted siRNA screen, we set out to address which actin nucleators play essential roles by monitoring the robust blebbing activity of cells during the process of cell adhesion. Our results identify the human formin DIAPH3 as a novel and essential factor involved in PM blebbing and implicate isoform-specific functions for this actin nucleator during cell protrusion.
Results
Adhering HeLa cells display dynamic PM blebbing
In search of an easily accessible experimental model system that would allow us to identify and validate relevant components for PM blebbing in human cells without genetic manipulation, we first examined whether HeLa cells undergo dynamic PM blebbing when adhering to uncoated plastic or glass surfaces as described for xenopus endothelial cells 19. To be able to monitor PM blebbing dynamics and F-actin remodeling in living cells, we generated HeLa cell derivatives that stably express the F-actin-binding probe Lifeact.GFP (Figure 1A) 27. 2 h post plating, more than 60% of these HeLa cells displayed prominent PM blebs with some incorporation of F-actin in the bleb rim (Figure 1A and Supplementary information, Figure S1B). Consistent with reports from other blebbing models 21, 25, PM blebbing during HeLa cell adhesion was highly dynamic, characterized by rapid bleb expansion followed by a static phase and subsequent slow retraction of the bleb (Figure 1A, see also Supplementary information, Movie S1). Moreover, F-actin was only detectable in blebs following the initial expansion phase, but remained associated with the bleb rim during bleb retraction (Figure 1A, lower panel). Addition of the actin polymerization inhibitor cytochalasin D (Cyto D) or the Rho-kinase (ROCK) inhibitor Y-27632 rapidly arrested blebbing (Figure 1B and Supplementary information, Movies S2 and S3). While Cyto D induced the formation of large static blebs devoid of F-actin at their rim, inhibition of ROCK triggered a gradual disappearance of blebs. In contrast, microtubule disruption by addition of nocodazole (Noco) increased blebbing to result in the formation of extended bleb-in-bleb structures (Figure 1B and Supplementary information, Movie S4). Consistent with the requirement for ROCK, PM blebbing of adhering HeLa cells strictly required the activity of the GTPase and ROCK regulator RhoA, while activity of Rac1 and Cdc42 GTPases was dispensable (Supplementary information, Figure S1A and S1B). Finally and in line with previous results on blebs observed upon SH4-domain expression or filamin A deficiency, PM blebs were enriched in p-MLC, p-ERM and p-Src that displayed distinct localizations at blebs (Supplementary information, Figure S1C) 12, 20. Together, these results establish PM blebbing of adhering HeLa cells as a model system that mirrors the general characteristics of PM blebbing and in which blebbing is governed by Rho-ROCK signaling and F-actin remodeling.

PM blebbing during HeLa cell adhesion depends on DIAPH3. Hela cells stably expressing the F-actin-binding probe Lifeact.GFP were analyzed by wide-field live-cell imaging 2-5 h post plating on uncoated plastic or glass surfaces. Scale bars, 10 μm. (A) Adhering HeLa cells display highly dynamic PM blebs. Still pictures of the indicated time points of Supplementary information, Movie S1 are shown. The bottom panel depicts enlarged images of areas indicated in the top panel by white boxes. Arrows point at individual sites of bleb formation and follow the individual blebs throughout their life cycle. Note that expanding blebs are initially devoid of F-actin. (B) Effects of cytoskeleton-disrupting drugs on PM blebbing of adhering HeLa cells. Shown are still pictures from the indicated time points of Supplementary information, Movies S2-S4. At time point 0 s, Cyto D (2 μM), Y-27632 (10 μM) or Noco (100 μM) were added. Arrowheads point to bleb-in-bleb structures observed after Noco treatment. (C-E) Small-scale RNAi screen for human actin nucleators. HeLa cells or Lifeact.GFP-expressing HeLa cells were transfected with 30 pmol of the indicated siRNA SMARTpools. 96 h post transfection, cells were replated and cell morphology and PM blebbing efficiency were analyzed on fixed cells stained for F-actin or by live-cell imaging of Lifeact.GFP-expressing cells. (C) Quantification of the percentage of cells that displayed pronounced PM blebbing following treatment with the indicated siRNAs. Presented are mean values ± SD from five independent screening experiments, except for JMY where the mean value of two independent experiments is shown. Blebbing efficiencies for DAAM2 and FMN2 are not shown since low basal mRNA levels precluded assessment of knockdown efficiencies (see Supplementary information, Figure S2 for summary of individual screening rounds). At least 100 cells were analyzed for each condition. Bars in grey indicate siRNA smartpools that induced statistically significant reduction in blebbing efficiency. (D) Western blots from lysates transfected with control siRNA or DIAPH1 and DIAPH3-specific siRNA SMARTpools, respectively. The dashed line indicates where the membrane was cut and incubated separately under identical conditions with DIAPH3-specific and secondary antibodies for detection of low abundant, faster migrating MW species. (E) Stills of the Supplementary information, Movies S5 and S6 are shown for control and DIAPH3 siRNA. Inset boxes represent magnified selected regions of cells, which are indicated by arrows. Note the lack of PM blebbing and the pronounced cell spreading in DIAPH3 siRNA-treated cells.
Screening for actin nucleators involved in PM blebbing
We next used this experimental system for a targeted siRNA screen to identify actin nucleators involved in PM blebbing during HeLa cell adhesion. Similar to a previous screen for the role of formins in cancer cell invasion 28, silencing against all 21 actin nucleators known to be expressed in human non-muscle cells was achieved by transfection with SMARTpool siRNAs consisting of four distinct siRNAs against each target. 96 h post transfection, cells were replated and the frequency of cells with prominent PM blebs quantified. A total of five independent rounds of screening was conducted (three on cells fixed post plating, two with live cell imaging of adhering cells, see Supplementary information, Figure S2A and Figure S2C for representative pictures and a summary of the results of each screening round). As shown in the summary of the results of all screening rounds (Figure 1C), PM blebbing of HeLa cells after treatment with a non-targeting control siRNA was observed in almost 80% of the cells. The majority of nucleators such as DIAPH2, FMNL1 or Arp3 appeared dispensable for PM blebbing, despite efficient knockdown at the mRNA level (see knockdown efficiencies in Supplementary information, Figure S2B). For DAAM2 and FMN2, knockdown efficiencies could not be determined due to their low mRNA levels in our HeLa cells thus precluding assessment of their relevance for PM blebbing (see Supplementary information Figure S2). In turn, silencing of expression of DIAPH1, DIAPH3, FMN1, Delphilin, IFN2 and Spir1 resulted in a statistically significant reduction of the percentage of blebbing cells. Among these six candidate actin nucleators involved in PM blebbing, silencing of DIAPH3 expression (see western blot shown in Figure 1D, lower panel) resulted in the most robust reduction of blebbing and caused pronounced morphological alterations of adhering cells. In addition to a complete abrogation of PM blebbing in many cells, reduction of DIAPH3 expression promoted cell spreading (Figure 1E and Supplementary information, Movies S5 and S6). As the current knowledge about this human formin is very limited, we decided to further examine the role of DIAPH3 (also referred to as Drf3) in PM blebbing.
Inverse correlation between PM blebbing and cell spreading
Triggered by the altered morphology of adhering HeLa cells following knockdown of DIAPH3, we addressed the relationship between PM blebbing and cell spreading on different substrates. Since cell spreading is strongly facilitated by engagement of integrins 29, fibronectin (Fbn) (Figure 2) and collagen I (data not shown) were employed to trigger integrin activation, yielding similar results. Expectedly, HeLa cells 3 h post plating on Fbn-coated surfaces displayed potent cell spreading in the virtual absence of PM blebs (Figure 2A, compare uncoated and Fbn). Kinetic analysis of frequency of cells with PM blebs revealed that on uncoated surfaces, blebbing was efficiently observed as early as 30 min post plating and that the blebbing frequency further increased over time. In contrast, efficient blebbing was not observed at any time of the observation period when cells were plated on Fbn-coated surfaces (Figure 2B, left panel). Consistent with a recent report 30, cell spreading, however, steadily increased with time on Fbn-coated surfaces, while blebbing cells on uncoated surfaces only displayed inefficient cell spreading (Figure 2B, right panel). To test if this switch between PM blebbing and cell spreading in the presence of Fbn was mediated by integrins, cells were incubated with the β1-integrin-blocking antibody 4B4 or with an antibody against transferrin receptor (TfR) as isotype control. While blebbing was not affected by treatment with a β1-integrin-blocking antibody on uncoated control surfaces, it was fully restored in the presence of the 4B4 antibody on Fbn-coated surfaces (Figure 2C and 2D). Similarly, silencing of β1-integrin expression resulted in efficient blebbing of HeLa cells on Fbn (Figure 2E and 2F). Thus, β1-integrin-mediated signaling and concomitant cell spreading negatively regulate PM blebbing of adhering cells, suggesting that the initiation of cell spreading by integrin activation may act as a stop signal for PM blebbing. Importantly, kinetic analysis of PM blebbing and cell spreading of HeLa cells in the absence of integrin activation by Fbn but following silencing of DIAPH3 revealed that blebbing was decreased and cell spreading enhanced, respectively, relative to the control siRNA (Figure 2G and 2H). This induction of cell spreading upon depletion of DIAPH3 was paralleled by the formation of elongated mature focal adhesions, while in blebbing control cells, small focal complexes were detected (Supplementary information, Figure S3) 31. Although less potent than Fbn, suppression of DIAPH3 thus appears to phenocopy integrin activation with regards to cell spreading and PM blebbing.

siDIAPH3 leads to decreased PM blebbing and enhanced spreading during HeLa cell adhesion, mimicking integrin engagement. All quantifications present mean values ± SD from three independent experiments. Over 100 or 25 cells were analyzed per experiment for determination of blebbing frequencies and adhesion areas, respectively. Scale bars, 10 μm. (A-B) HeLa cells were plated on uncoated or Fbn-coated coverglasses and fixed at different time points after plating. (A) Confocal microscopy images of cells stained for F-actin at 3 h post plating. (B) Quantification of blebbing frequencies (left panel) and cell adhesion areas (right panel). (C-D) Treatment of HeLa cells with the β1-integrin-blocking antibody 4B4 induces PM blebbing on Fbn-coated surfaces. Cells were pretreated with 4B4 antibody for 30 min, detached and replated in the presence of the 4B4 antibody onto uncoated or Fbn-coated coverglasses for 2 h. (C) Confocal images of cells stained for F-actin and (D) quantification of blebbing frequencies are shown. Cells without antibody addition or following incubation with an isotype control antibody were used as controls; TfR, transferrin receptor. (E-F) Silencing of β1-integrin expression induces PM blebbing on Fbn-coated surfaces. (E) Confocal images of cells stained for F-actin and (F) quantification of blebbing frequencies are shown. The inset in F depicts a western blot from lysates of the cells used for analysis of blebbing. (G-H) Analysis of HeLa cells treated with DIAPH3-specific or control siRNA. Cells were plated on uncoated coverglasses 96 h post siRNA transfection and analyzed for (G) morphology, (H) blebbing frequency (left panel) and adhesion area (right panel) in analogy to panels (A-B).
PM blebbing involves a specific DIAPH3 isoform
We next sought to rescue PM blebbing in DIAPH3-depleted cells by re-introduction of DIAPH3. Interestingly, mRNA and/or protein sequences of multiple isoforms of DIAPH3 generated by alternative splicing are reported in the NCBI and Swiss-Prot databases (see schematic in Figure 3A). To address which of these isoforms are expressed in our HeLa cells, we first analyzed the presence of DIAPH3-specific mRNA species by northern hybridization (Figure 3B). Indeed, a complex but very similar pattern was observed in total RNA of HeLa as well as 293T cells, suggesting that multiple DIAPH3 isoforms are indeed expressed in human cells. RNA from murine NIH3T3 cells did not give rise to any specific signal (Figure 3B). The dominant signal in the human cell lines corresponded well to the expected size of the longest DIAPH3 isoform 3 mRNA (4.8 kb+polyA, marked by an asterisk). Alternative splice products were present in lower abundance, however, one of those was concordant in size to the mRNA of isoform 1 (2.7 kb + polyA, marked by a dot), indicating that this isoform is present in HeLa cells. Consistently, at least four different DIAPH3 species were detected by western blotting in HeLa cell lysates (Figure 3C and 3E), all of which were affected by the pool of siRNAs used in our screen (Figure 1D). Among these, a high molecular weight species was by far most abundant (Figure 1D). Shorter exposure of the western-blot membrane revealed the existence of two distinct high molecular weight DIAPH3 species (Figure 3C lane (−)). Based on their comigration with ectopically expressed variants of the respective isoforms and confirmation of their corresponding 3′ ends at the mRNA level (data not shown, see also Supplementary information, Figure S4A), these DIAPH3 species were identified as isoform 3 (Isof3) and isoform 7 (Isof7). Similarly, a lower molecular weight species of significantly lower abundance was identified as isoform 1 (Isof1) (Figure 3D). Of note, endogenous expression of Isof3, Isof7 and Isof1 was detected in Hela and 293T cells (Figure 3E). The identity of the remaining molecular weight species remained unclear.

Several distinct DIAPH3 isoforms are expressed by HeLa cells. (A) Schematic representation of DIAPH3 isoforms produced by alternative splicing; GBD, GTPase-binding domain; FH, formin homology domain; DID, diaphanous inhibitory domain; DAD, diaphanous autoregulatory domain. Isoform identification and domain numbering are based on entries in the Swiss-Prot database, Swiss-Prot identifiers are shown in brackets. Isoform 3 represents the canonical sequence, amino-acid differences in other isoforms are depicted in black. Note that isoform 3 and isoform 1 are identical to the major transcript variants deposited in the NCBI nucleotide database, referred to as isoform a (accession number NM_001042517.1/NP_001035982.1) and isoform b (accession number NM_030932.3/NP_112194.2). Positions of the negative sense riboprobe used in B and of the DIAPH3 antigen used to generate the DIAPH3 antibody employed for detection of endogenous DIAPH3 are indicated by black and grey lines, respectively. (B) Analysis of DIAPH3 mRNAs in different cell lines. 10 μg RNA isolated from HeLa, NIH3T3 or 293T cells were subjected to northern hybridization using a DIAPH3-specific riboprobe complementary to a part of the coding region as indicated in A. Increasing amounts of in vitro transcripts encompassing the complete coding sequence of isoform 1 were used as a positive control. The positions of 28S RNA (4.7 kb) and the size of the control in vitro transcript are depicted on the right. The band matching to the expected size of isoform 3 (4.8 kb, NM_001042517.1) in HeLa cells is marked with an asterisk, a potential isoform 1 (2.7 kb, NM_030932.3) mRNA is marked by a dot. (C-E) Western blot analysis for identification of DIAPH3 isoforms on the protein level. (C) Lysates of 293T cells expressing HA.DIAPH3 Isof3 or Isof7 and lysates of untransfected HeLa cells (-) were probed with DIAPH3- (left lanes) or HA-specific (right lanes) antibodies. Dots indicate the position were the membrane was cut. (D) Western blot of lysates of 293T cell lysates expressing HA.DIAPH3 Isof1 or untransfected HeLa cell lysates. The dashed line indicates where the membrane was cut and incubated separately under identical conditions with DIAPH3-specific and secondary antibodies for detection of low abundant, faster migrating MW species. (E) Western blot of lysates of untransfected HeLa and 293T cells analogous to D.
Attempts to specifically interfere with the expression of individual DIAPH3 isoforms via sequence- or UTR-specific siRNAi remained unsuccessful and indicated that the stability of individual DIAPH3 isoforms might depend on the presence of all other isoforms (data not shown). We thus generated expression constructs for DIAPH3 Isof1, Isof3 and Isof7 as GFP fusion proteins to address their relevance for PM blebbing. Although being an abundant DIAPH3 isoform expressed in our HeLa cells, DIAPH3 Isof3 (aa 1-1193; GFP.Isof3) did not alter the blebbing behavior of control cells and did not result in a significant increase of PM blebbing in HeLa cells treated with DIAPH3-specific siRNA (Figure 4A and 4B). Similarly, no role in PM blebbing was observed for DIAPH3 Isof7 (GFP.Isof7) that encompasses aa 1-1112 and was previously referred to as full-length mDia2/Drf3 32. We therefore tested a variant of Isof7 that is artificially activated by deletion of its autoinhibitory DAD domain (GFP.Isof7ΔDAD), but is unlikely to be expressed endogenously. Consistent with previous reports for DIAPH3 as well as for analogous constructs of the DIAPH3 mouse ortholog mDia2 32, 33, 34, expression of Isof7ΔDAD resulted in the formation of multiple filopodia but not blebs independently of whether or not endogenous DIAPH3 was depleted. In sharp contrast, expression of isoform 1 fused to GFP (GFP.Isof1, encompassing residues 264-1112) in DIAPH3 knockdown cells induced efficient PM blebbing that was sensitive to inhibition of ROCK (Figure 4A and B, data not shown). These cells also displayed occasional F-actin fibers (data not shown). In contrast to the frequency with which PM blebbing was observed, the number of blebs per individual cell was not increased by GFP.Isof1 expression (Figure 4C). Even though individual blebs induced by GFP.Isof1 were morphologically comparable to blebs of control cells, they were distributed predominantly dorsally and failed to cover the entire cell surface including the cell periphery (Figure 4D). These blebs were overall less dynamic than typical blebs during cell adhesion but during their formation, dynamic blebs displayed the characteristic incorporation of F-actin at late stages of the bleb cycle (Figure 4E). These results suggest that the different DIAPH3 isoforms expressed in human cells exert individual functions and that PM blebbing of adhering HeLa cells specifically involves the DIAPH3 isoform 1. In this scenario, DIAPH3 Isof1 is in principle sufficient to cause PM blebbing during HeLa cell adhesion; however, additional factors are likely involved in the coordination of the dynamic blebbing process.

A specific isoform of DIAPH3 mediates PM blebbing during HeLa cell adhesion. (A-C) HeLa cells were transfected with control or DIAPH3-specific siRNA. Transfections with GFP.DIAPH3-encoding plasmids were performed 48 h post siRNA treatment. 24 h post plasmid transfection, cells were replated on uncoated surfaces for 3.5 h. (A) Wide-field microscope images of cells stained for F-actin (red) as well as GFP fluorescence (green) are shown. Scale bars, 10 μm. Right panels depict enlarged merged pictures of areas indicated by white boxes. Scale bars, 2.5 μm. (B) Quantification of the frequency of transfected cells that display blebs or filopodia. Data are mean ± SD from three independent experiments with at least 100 cells analyzed per experiment. (C) Quantification of bleb number per cell. Z-stacks of individual cells transfected with siRNA and plasmid DNA as indicated were acquired and blebs were counted manually. (D) HeLa cells were transfected with the indicated expression constructs. 24 h post transfection, cells were replated on uncoated surfaces for 3.5 h. Z-stacks of fixed cells stained for F-actin were acquired by spinning disk microscopy. Z-stacks were combined and the coronal and sagittal planes as indicated by white lines as well as maximum projection images are shown. Scale bars, 10 μm. (E) HeLa cells stably expressing Lifeact.GFP were transfected with an expression construct for mCherry.DIAPH3 Isof1. 24 h post transfection, cells were replated on uncoated glass surfaces and analyzed by wide-field live cell imaging 3.5 h post plating. Still pictures of Lifeact.GFP in a cell expressing mCherry.DIAPH3 Isof1 (mCherry fluorescence not shown) are depicted at the indicated time points of the movie. Scale bar, 10 μm. The bottom panel shows enlarged images as indicated in the top panel by white boxes. Arrow points at a dynamic bleb that incorporates F-actin in its rim at later stages of bleb life cycle. Scale bar, 5 μm.
Dimerization and actin nucleation are critical for DIAPH3-mediated PM blebbing
To explore the mechanism by which DIAPH3 facilitates PM blebbing, we expressed a series of DIAPH3 variants in HeLa cells (see western blot analysis in Figure 5C, bottom panel) and analyzed them at steady state 24 h post transfection. Expression levels varied between these constructs, which however did not affect our single-cell analysis that only considered cells with comparable intensity of GFP fluorescence. GFP.Isof3 and GFP.Isof7 localized diffusely in the cell cytoplasm and did not cause formation of cell protrusions or affect cellular F-actin organization (Figure 5B and 5C). Expression of GFP.Isof7ΔDAD frequently induced the potent formation of filopodia and the formin localized to the tips of these protrusions as well as to the PM in general. In contrast, GFP.Isof1 triggered PM blebbing with the formin localizing to the bleb lumen. An alanine-to-valine polymorphism at position 274 of Isof1 did not affect its subcellular localization or ability to induce PM blebbing. Single alanine substitutions of residues W651 or I725 in the FH2 domain, which are critical in the murine ortholog of DIAPH3, mDia2, for dimerization or actin nucleation 35, 36 (see schematic in Figure 5A), respectively, fully abrogated bleb induction by Isof1. Both residues were also essential for filopodia formation by Isof7ΔDAD even though these mutant proteins still associated with the PM. Similar results were obtained when PM blebbing was observed during HeLa cell adhesion 3.5 h post plating (Supplementary information, Figure S5). This analysis also indicates that the induction of filopodia occurs in the absence of bleb formation in adhering cells.

Specific DIAPH3 variants selectively induce PM blebs or filopodia in a dimerization and actin nucleation-dependent manner. (A) Schematic representation of the DIAPH3 constructs used. Amino-acid numbering and domain layout is based on the Swiss-Prot database. GBD, GTPase-binding domain; FH, formin homology domain; DID, diaphanous inhibitory domain; DAD, diaphanous autoregulatory domain. Point mutations are indicated by black bars, W651A and I725A designates DIAPH3 mutant proteins deficient in dimerization and actin nucleation, respectively. The right panel summarizes the effects of the various DIAPH3 constructs on blebs and filopodia induction as well as their subcellular localization. (B-C) HeLa cells were transfected with expression constructs for the indicated GFP.DIAPH3 fusion proteins and fixed 24 h post transfection. (B) Confocal images of cells stained for F-actin (red) as well as GFP fluorescence (green) are shown. Scale bars, 10 μm. The bottom panel depicts enlarged merge pictures of the areas indicated in the top panel by white boxes. Scale bars, 5 μm. (C) Quantification of the frequency of transfected cells that display blebs or filopodia. Data are mean ± SD from three independent experiments with at least 100 cells analyzed per experiment. The bottom panel depicts a western blot from lysates of cells transfected with the indicated expression constructs. (D) Quantification of SRE transcription. NIH3T3 cells were co-transfected with pTK-Renilla, a 5× SRE-Luc reporter plasmid and the indicated GFP.DIAPH3 expression construct and lysed 24 h post transfection. Luciferase activity was measured and normalized for transfection efficiency. Fold activation was calculated relative to samples transfected with the pEGFP-C1 vector control. Data are mean ± SEM from three independent experiments performed in triplicates.
Since transcription from the serum response element (SRE) represents an established effector function of actin remodeling triggered by diaphanous formins including mouse mDia2 37, 38, we assessed effects of DIAPH3 isoforms and mutants on SRE activation using a SRE-luciferase reporter construct (Figure 5D). In line with the observed absence of actin remodeling, GFP.Isof7 failed to induce significant activation of serum response factor (SRF) transcriptional activity. In contrast, both GFP.Isof7ΔDAD and GFP.Isof1 triggered a potent SRF response, demonstrating that SRF activation is not affected by the type of cell protrusion induced by the respective DIAPH3 variant. Notably and confirming the specificity of the I725A mutation in the context of DIAPH3, the corresponding FH2 domain mutants lacked any detectable SRF activation. Together, these results demonstrate that bleb and filopodia-inducing DIAPH3 variants differ in their subcellular localization while they both depend on dimerization and actin nucleation for induction of the respective cell protrusions as well as for SRF activation.
DIAPH3 has potent actin assembly activity
Next, we assessed whether human DIAPH3 indeed functions as an actin nucleation and assembly factor. To this end, FH2-DAD and FH1-FH2-DAD fragments of DIAPH3 were recombinantly expressed and purified (Figure 6A and 6B). In vitro analysis of F-actin assembly kinetics of pyrene-actin revealed that the FH2-DAD fragment of DIAPH3 is sufficient to polymerize actin (Figure 6C) and that the additional presence of the FH1 domain does not significantly affect this activity (Figure 6D). Magnitude and kinetics of actin assembly were comparable to the corresponding fragment of the mouse counterpart mDia2 (Figure 6E). These results reveal the human formin DIAPH3 as a potent actin assembly factor.

DIAPH3 exerts potent actin nucleation activity. (A-B) Purified His.DIAPH3 fragments were separated by 12% SDS-PAGE followed by (A) Coomassie staining and (B) western blotting using a DIAPH3-specific antibody. (C-E) Pyrenyl actin assembly assays were performed with 2 μM actin, 10% pyrene labeled, and respective DIAPH3 or mDia2 fragments as indicated. (C) DIAPH3 FH2/DAD, (D) DIAPH3 FH1/FH2/DAD, (E) mDia2 FH1/FH2/DAD. Actin was assembled in control buffer or in the presence of different concentrations of individual proteins as indicated. Actin assembly activity is displayed in fluorescence arbitrary units (AU). FH, formin homology domain; DAD, diaphanous autoregulatory domain.
The GBD determines the cell protrusion specificity of DIAPH3 variants
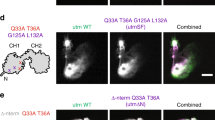
We then sought to define the molecular determinants that govern the specificity of Isof1 or Isof7ΔDAD for the formation of PM blebs or filopodia, respectively. To this end, we constructed a series of DIAPH3 deletion mutants (schematic in Figure 7A) that were expressed to somewhat variable levels as GFP fusion proteins in HeLa cells (Figure 7C, bottom panel). Of note, the filopodia-inducing DIAPH3 variant GFP.Isof7ΔDAD lacks a C-terminal DAD domain present in the bleb-inducing isoform 1. Deletion of the DAD (see Isof1ΔDAD), however, did not affect induction of PM blebbing or localization of this protein to the bleb lumen (Figure 7B and 7C). However, the frequency of blebbing was slightly reduced in all bleb-inducing DIAPH3 variants that lack a DAD (compare e.g., Isof1 and Isof1ΔDAD). Thus, although the DAD is not essential for blebbing per se, it contributes to efficient bleb induction via DIAPH3.

The GTPase-binding domain determines the protrusion specificity of individual DIAPH3 variants. (A) Schematic representation of DIAPH3 constructs used. Amino-acid numbering and domain layout is based on the Swiss-Prot database. GBD, GTPase-binding domain; FH, formin homology domain; DID, diaphanous inhibitory domain; DAD, diaphanous autoregulatory domain. Effects on blebs and filopodia induction by distinct constructs and construct localization are summarized in the right panel. (B-C) HeLa cells were transfected with the indicated GFP.DIAPH3 expression contsructs and fixed 24 h post transfection. (B) Confocal images of cells stained for F-actin (red) as well as GFP fluorescence (green) are shown. The bottom panel depicts enlarged merge pictures of the areas indicated in the top panel by white boxes. Scale bars, 5 μm. (C) Quantification of the frequency of transfected cells that display blebs, filopodia or both types of protrusions. Data are mean ± SD from four independent experiments with at least 100 cells analyzed per experiment. The bottom panel depicts a western blot from lysates of cells transfected with the indicated expression constructs.
The filopodia-inducing DIAPH3 proteins all contain N-terminal regions that have been implicated in membrane targeting of the mDia1 39. However, deletion of the first 113 residues of DIAPH3 Isof7ΔDAD reported to mediate membrane contacts in mDia2 (see 114-1057) did not affect filopodia induction and tip localization (Figure 7). Furthermore, addition of the putative membrane-targeting signal to the bleb-inducing DIAPH3 versions (1-113/264-1112 and 1-113/264-1057) did not affect bleb induction and also failed to target DIAPH3 to cellular membranes (Figure 7). Therefore, the N-terminal 113 amino acids of DIAPH3 are unlikely to determine membrane association or protrusion specificity of DIAPH3.
In contrast, further N-terminal deletions into the GBD of the filopodia-inducing Isof7ΔDAD shifted DIAPH3′s biological properties and turned the protein into a bleb-inducing variant that was now recruited to the bleb-lumen. This phenotype was induced already by small truncations (151-1057) and persisted upon further deletion into the GBD/FH3 region (Isof7ΔDADΔGBD). These findings implicate a central role for the GBD/FH3 region in determining protrusion specificity, which was further supported when analyzing blebbing in DIAPH3-expressing HeLa cells 3.5 h post plating (Supplementary information, Figure S6). Of note, introduction of a H181D mutation known to disrupt the interaction between the mDia2-GBD and Cdc42 34, 40, did not affect filopodia induction and failed to promote bleb formation by Isof7ΔDAD (see Isof7ΔDAD H181D, Figure 7B). Together, these results reveal the GBD as a key determinant for subcellular targeting and protrusion specificity of activated human DIAPH3 and further define isoform 1 as a non-GTPase-regulated protein that is specifically involved in PM blebbing.
Discussion
PM blebbing is observed under distinct physiological scenarios triggered by a variety of extracellular cues. Despite this diversity, bleb dynamics follows common principles and relies on Rho-ROCK-mediated contractility. Only few studies previously addressed the involvement of specific actin nucleators in PM blebbing. Using experimental systems in which PM blebbing was induced by genetic manipulation or protein overexpression, nucleators such as mDia1, mDia2, FHOD1, Arp2/3 that are also involved in the regulation of diverse additional cellular actin remodeling events were implicated in blebbing 12, 41, 42, 43. In this present study, we conducted a comprehensive analysis of the involvement of endogenously expressed human actin nucleators in PM blebbing of adhering HeLa cells. siRNA screening identified six nucleators with essential roles in PM blebbing. Even though overexpression of individual nucleators may be sufficient for the induction of blebbing, these results suggest that physiologically, several nucleators may cooperate to coordinate PM blebbing. Conceivably, the set of nucleators involved may differ between cell types and bleb stimuli. Identified already as a prerequisite for PM blebbing during amoeboid invasion of cancer cells 41, Dia1 may represent a nucleator with roles in a broad range of PM blebbing scenarios. Interestingly, with Spir1 and FMN1, we also identified two nucleators that cooperate in regulated actin remodeling 44, 45, and addressing their specific role in PM blebbing will be an interesting goal for future studies.
Since the human formin DIAPH3 is not very well studied, we decided to investigate its potential involvement in bleb protrusions further. Partial inhibition of DIAPH3 expression not only efficiently reduced PM blebbing but also triggered rapid cell spreading. The murine ortholog of DIAPH3, mDia2, also exerts well-established roles in diverse cellular processes such as formation of filopodia or lamellipodia and completion of cytokinesis 32, 33, 34, 37, 46, 47. Our results suggest that distinct DIAPH3 isoforms, generated by alternative splicing, govern specific biological processes as an important source for such apparent multifunctionality. Since cell type- or organelle-specific isoforms were also identified for hDia2C and FHOD3 48, 49, human cells likely display a significantly higher complexity of actin nucleator proteins than currently appreciated. Conceivably, cells likely adjust the composition and regulation of this pool of actin-remodeling factors to meet specific needs.
In the case of HeLa cells, ectopic expression of DIAPH3 Isof1 induced blebbing irrespective of whether or not expression of endogenous DIAPH3 was silenced. While this identifies Isof1 as sufficient for the induction of PM blebbing, bleb number, position and dynamics were not fully restored upon exclusive expression of Isof1. Consistent with the fact that our various siRNA approaches all induced silencing of several DIAPH3 isoforms in addition to Isof1, these results indicate that different DIAPH3 isoforms may cooperate in the coordination of dynamic PM blebbing. Alternatively, the elevated per cell levels of Isof1 following ectopic expression relative to those observed for endogenous Isof1 may interfere with the proper organization of blebbing. In addition to the identification of DIAPH3 Isof1 as a novel regulator of PM blebbing, our results confirmed that a constitutive active version of DIAPH3 Isof7 (Isof7ΔDAD) triggers formation of filopodia. Despite these different protrusion specificities, introduction of specific mutations that interfere with dimerization and/or actin nucleation by the FH2 domain completely inhibited generation of cell protrusions as well as SRF activation by both DIAPH3 variants. Together with the demonstration of potent in vitro actin nucleation by the FH1-FH2-DAD DIAPH3 module, these results reveal that DIAPH3 exerts classical formin functions. However, subcellular localization and biological outcome of the respective actin assembly activity of different isoforms were strikingly different. Truncation mutagenesis revealed that the N-terminus of Isof7, which is lacking in the bleb-inducing Isof1, is responsible for this functional difference. Since filopodia induction correlated with PM localization of the formin, we analyzed whether a putative membrane-targeting region in the far N-terminus was essential for this activity 39. However and in agreement with a recent study on mDia2 50, we failed to detect a significant contribution of the N-terminus of DIAPH3 for filopodia formation and membrane recruitment. Instead, we found that the GBD of Isof7ΔDAD was crucial for membrane association as well as for filopodia induction. Moreover, deletion of the GBD from DIAPH3 Isof7 switched this isoform from a membrane-associated promoter of filopodia formation to a cytoplasmic inducer of blebbing. Since the GBD was fully preserved in the context of a DAD deletion and thus absence of intramolecular autoinhibition of DIAPH3, we conclude that this region likely exerts additional functions beyond GTPase binding and regulation of autoinhibition. Our results strongly suggest that subcellular targeting, in the case of filopodia formation by DIAPH3 to the PM, represents such an activity. Given its similarity to Isof7, the activity of DIAPH3 Isof3 might be regulated by identical mechanisms.
Surprisingly, our results with DAD-deleted versions of Isof1 revealed a contribution of this domain to the efficiency of PM blebbing. In contrast to the classical role in autoinhibition of diaphanous formins, however, deletion of the DAD slightly decreased rather than stimulated the biological activity of DIAPH3. While this provides another indication for the lack of autoinhibition in DIAPH3 Isof1, this also suggests that the DAD may play an active role in DIAPH3 actin remodeling. Such an activity could be related to the potential ability of the DAD to regulate the activity of other formins in trans 38 or to recruit factors that facilitate DIAPH3-mediated actin remodeling.
Together, our results can be integrated into the following model to explain how different DIAPH3 isoforms are selectively involved in the formation of specific cell protrusions. DIAPH3 Isof7 is typically present in its autoinhibited form in the cytoplasm of cells. Engagement of its GBD will recruit the formin to the PM and release its autoinhibition. This recruitment may rely exclusively on the interaction with GTPases such as Cdc42 or Rif and may also involve yet-to-be defined alternative interactions of the GBD. Once localized to the PM in its active form, FH2-mediated actin assembly drives formation of filopodia with the formin localizing to the filopodia tip. In contrast, DIAPH3 Isof1 resides in the cell cytoplasm in an active form lacking autoinhibition. Subsequent to yet unknown regulation that may involve subcellular targeting and/or post-translational modifications but not GTPase-mediated activation, actin nucleation by this DIAPH3 isoform promotes dynamic PM blebbing. Since PM blebbing in our experimental system is only observed at early time points of cell adhesion and subsequently diminishes in spreading cells, this process must be negatively regulated by extracellular cues. Our results suggest that integrin engagement can provide such a signal to blunt PM blebbing and to promote cell spreading. The precise molecular mechanism of this regulation clearly requires further investigation but likely involves termination of stimulatory signals or modulation of DIAPH3 by the integrin cascade.
In sum, we identify PM blebbing and filopodia formation as specific functions that can be exerted by the DIAPH3 isoform 1 or isoforms 7 and 3, respectively and reveal that subcellular targeting by the GBD determines the specific biological outcome of DIAPH3-mediated actin assembly. The presence of actin nucleator isoforms with specific biological properties expands the plasticity of human cells for dynamic actin rearrangements in response to physiological cues.
Material and Methods
Expression constructs, reagents and antibodies
DIAPH3 cDNAs (NM_030932.3 and BC034952.1) were PCR amplified from Ultimate™ ORFs IOH26769 and IOH62076, respectively, in Gateway entry vectors (Invitrogen) and cloned into EcoRI/KpnI sites of pEGFP-C1. NM_030932.3 and BC034952.1 correspond to the DIAPH3 isoform 1 or its A274V polymorphism, respectively. EGFP-Drf3 full-length (DIAPH3 isoform 7) and EGFP-Drf3 delta DAD (DIAPH3 isoform 7ΔDAD) were gifts from J Faix 32. The EGFP-tagged DIAPH3 Isof3 was generated by ligation of the fragments 1-3 318 bp (amplified from the EGFP-Drf3 full-length construct) and 3 319-3 582 bp (amplified from HeLa cDNA) into the XhoI/KpnI sites of pEGFP-C1. The HA-tagged versions of DIAPH3 Isof1, Isof3 and Isof7 were constructed by PCR amplification of the individual isoforms with forward primers containing the HA-tag coding sequence and ligated into the AgeI/KpnI sites of pEGFP-C1. mCherry.DIAPH3 Isof1 was generated by EcoRI/KpnI digestion of the pEGFP-C1.DIAPH3 Isof1 plasmid and subsequent ligation of the released insert into pmCherry-C1. Dimerization and actin nucleation-deficient mutants were generated in analogy to mouse mDia2 35 by site-directed mutagenesis using the DIAPH3 expression constructs for Isof7, Isof7ΔDAD, Isof1 and Isof1 A274V as template, respectively. The Cdc42-binding mutant of DIAPH3 isoform 7ΔDAD (Isof7ΔDAD H181D) was generated in analogy to mouse mDia2 34 by site-directed mutagenesis of DIAPH3 isoform 7ΔDAD construct. N- and C-terminal DIAPH3 deletion constructs were generated by PCR amplification and were cloned into pEGFP-C1. The 1-113/264-1112 DIAPH3 construct was generated by ligation of two distinct PCR fragments into pEGFP-C1. Expression constructs for GFP-tagged C3 transferase and for Myc-tagged GTPases were described earlier 12. Transfection of plasmid DNA was performed using Lipofectamine 2000 (Invitrogen).
Reagents were as follows: Alexa Phalloidin-TRITC (Sigma), Phalloidin-FITC (Sigma), Cyto D (Sigma), Y-27632 (Calbiochem), Noco (Calbiochem), Mowiol 4-88 (Sigma) and Fbn (Sigma). 2.5 μg Fbn per cm2 was used for coating.
Antibodies used were as follows: rabbit anti DIAPH3 (ProteinTech Group), rabbit anti DIAPH3 (Sigma-Aldrich), rabbit anti phospho-myosin light chain 2 (Ser19) (Cell Signaling), rabbit anti phospho-Src (Tyr416) (Cell Signaling), rabbit anti phospho-Ezrin (Thr567)/Radixin (Thr564)/Moesin (Thr558) (Cell Signaling), rabbit anti HA Y-11 (Santa Cruz), mouse anti c-Myc 9E10 (Santa Cruz), mouse anti α-tubulin clone B-5-1-2 (Sigma), mouse anti β1-integrin (BD Biosciences), mouse anti-human TfR (Zymed Laboratories), mouse anti β1-integrin 4B4 (Beckman Coulter), mouse anti vinculin (Sigma-Aldrich), mouse anti GFP (Sigma), goat anti-rabbit Alexa Fluor 488 (Invitrogen), biotin-rat anti-mouse (Invitrogen), Cy3-conjugated streptavidin (Jackson ImmunoResearch).
Cell lines and culture
HeLa cells/HeLa cells stably expressing Lifeact.GFP and 293T cells were cultivated in DMEM medium with 4.5 g/l glucose, NIH3T3 cells in DMEM medium with 1.0 g/l glucose (Invitrogen) supplemented with 10% fetal calf serum (FCS) and antibiotics. For stable expression of Lifeact.GFP in HeLa cells, the respective coding sequence was cloned into BamHI and SmaI restriction sites of pWPXL (Addgene) from a Lifeact encoding pEGFP-N1 plasmid provided by R Wedlich-Söldner 27. Subsequently, pWPXL/Lifeact.GFP was co-transfected with psPAX2 (Addgene) and VSV-G-expressing plasmids into 293T cells and virus was harvested 72 h post transfection. Spin infection of HeLa cells was performed in presence of Polybrene (Sigma) and HeLa cell clones with stable Lifeact.GFP expression were selected by FACS sorting.
Immunostainings and microscopy
To detach HeLa cells, 5mM EDTA was applied and cells were subsequently replated into new dishes containing coverglasses for 3.5 h (or different time points where indicated). For F-actin staining, cells grown on coverglasses were fixed with 8% paraformaldehyde for 15 min, washed with PBS, permeabilized with 0.1% Triton X-100 and incubated with fluorescently conjugated Phalloidin at room temperature for 1 h. For antibody staining, cells were fixed and permeabilized, blocked with 1% BSA in PBS for 1 h and then stained with appropriate primary and secondary antibodies (room temperature, 1 h). For phospho antibodies, TBS was used instead of PBS and staining with primary antibody was performed at room temperature for 3-4 h. For vinculin staining, a biotin-conjugated secondary antibody was used followed by a staining with conjugated streptavidin for signal enhancement. Coverglasses were mounted in Mowiol. For live-cell imaging with subsequent drug addition, HeLa cells stably expressing Lifeact.GFP were replated into μ-Slides 8-well (Ibidi). Individual drugs or solvent were added during the imaging at final concentrations: 2 μM Cyto D, 10 μM Y-27632 and 100 μM Noco, respectively. Cells were quantified, imaged and live-cell microscopy of drug-treated cells was performed using an Olympus IX81 microscope with cellM software. Live-cell microscopy of HeLa cells stably expressing Lifeact GFP during the siRNA screen was performed on a Zeiss Axiovert 200 M microscope with Metamorph Software equipped with incubation chamber. Confocal images were taken using Zeiss LSM 510 Axiovert, Leica TCS SP5 or Perkin Elmer spinning disk confocal ERS6-line (Nikon Imaging Center Heidelberg) microscopes. The presence of blebs was assessed microscopically in the F-actin channel and cells were scored positive for blebbing if three or more well-visible blebs protruded from their cell surface.
siRNA knockdowns
For siRNA screen-mediated knockdowns, cells were transfected with 30 pmol of SMARTpool siRNA, i.e., four oligonucleotides targeting distinct sites in RNA (Dharmacon). See Supplementary information, Figure S7 for catalog numbers and target GENE IDs for formin nucleators and Supplementary information, Figure S8 for the target sequences of the DIAPH3 smartpool in diaph3 isoforms. For hDia2C, the siRNA sequence 5′-ucuauaguuggaaguaaagtt-3′ was used. Unspecific control siRNA (MWG) sequence: 5′-agguaguguaaucgccuugtt-3′. Briefly, siRNA was diluted in Opti-MEM I medium (Invitrogen), Lipofectamine RNAiMAX (Invitrogen) was added and the mixture was incubated at room temperature before being added to diluted cells in complete medium seeded simultaneously into 6-well plates. Cells were incubated without medium change. Cells were replated on uncoated coverglasses for analysis of fixed cells or on Glass Bottom Microwell Dishes (MatTek Corporation) for live-cell imaging 96 h post siRNA transfection. For adhesion area measurement/vinculin staining and siRNA rescue experiments, cells were transfected with a single siRNA derived from the DIAPH3 SMARTpool, target sequence: 5′-guagacauuugcauagauc-3′. For siRNA rescue experiments, GFP.DIAPH3 expression constructs were transfected 48 h post siRNA transfection and cells were replated on uncoated coverglasses 24 h post plasmid transfection. Knockdown efficiencies were determined by qPCR. Total RNA was isolated using Trizol (Invitrogen) followed by chloroform extraction, precipitated with isopropanol and dissolved in water (Merck). RNA was treated with TURBO DNase (Applied Biosystems) and reverse transcribed using M-MuLV reverse transcriptase (BioLabs). qPCR was performed with 7500 System Software (Applied Biosystems) using Power SYBR Green PCR Master Mix (Applied Biosystems). Primers used for qPCR are summarized in Supplementary information, Figure S9 and were partially validated previously 28. The DIAPH3 amplicon is between 3154-3255 bp of DIAPH3 Isof3 and is thus present within all putative isoforms except for Isof2. For β1-integrin knockdown, a validated siRNA from Qiagen was used (Hs_ITGB1_5 FlexiTube siRNA; SI00300573) and the knockdown efficiency was determined by western blotting.
Northern blotting
Northern blot analysis and in vitro transcription was performed as described previously 51. In brief, total RNA was isolated from HeLa, NIH3T3 and 293T cells using Trizol following the instructions of the manufacturer (Invitrogen). 10 μg of total RNA was denatured by treatment with 5.9% glyoxal in 50% dimethylsulfoxide and 10 mM sodium phosphate buffer, pH 7.0 separated by electrophoresis on a 1% agarose gel in 10 mM sodium phosphate buffer, pH 7.0 and transferred to a positively charged nylon membrane in 0.1 M NaOH using a vacuum blotting device. Prior to hybridization, the membrane was stained with methylene blue to localize the 28S rRNA. Hybridization followed standard techniques 52 using a 32P-labelled negative sense riboprobe for detection of mRNAs encoding DIAPH3 isoforms.
To generate a template for in vitro transcripts corresponding to the coding sequence of DIAPH3 isoform 1, the gene was amplified by PCR and fused to a T7-promoter using forward primer 5′-ttgtaatacgactcactatagggatgagtgaggagaggagcct-3′ and reverse primer 5′-ttataaatacggtttattaccatgg-3′ on plasmid template pEGFP-C1/DIAPH3 isoform 1 (264-1 112). For detection of differentially spliced DIAPH3 transcripts, a region encompassing 1.1 kb of the 5′-coding region of Isof1 (corresponding to 5-1 111 bp of DIAPH3 isoform 1) was amplified using forward primer 5′-gtgaggagaggagcctttcc-3′ and reverse primer 5′-ttgtaatacgactcactatagggacccaaatggcaggattggta-3′ to generate a template suitable for negative strand probe synthesis. All PCR products were purified by phenol/chloroform extraction. In vitro transcription and purification of transcripts was done as described 51.
In vitro actin nucleation assay
Human DIAPH3 FH2/DAD (amino acids 616-1093), DIAPH3 FH1/FH2/DAD (amino acids 561-1 093), and mouse mDia2 FH1/FH2/DAD (amino acids 534-1171) cDNA fragments were cloned into the pET20 + 2× His vector. Proteins were expressed in the DE3 E. coli strain at 25 °C for 4 h and purified using NiNTA agarose beads (Qiagen) and polypropylene columns (Qiagen). Proteins were dialyzed against 2 mM NaH2PO4 pH7, 150 mM NaCl, 0.1 mM EGTA, 0.1 mM MgCl2, 0.5 mM DTT. Pyrenyl actin assembly assays were performed with minor modifications, as described 54 using a reaction mix containing 10 μl energy-regenerating mix (150 mM creatine phosphate, 20 mM ATP, 2 mM EGTA, 20 mM MgCl2), 10 μl pyrene-labeled actin (2 μM final concentration, 10% labeled; Hypermol) and proteins at required amounts. Pyrene fluorescence was measured at 407 nm with excitation at 365 nm, and the kinetics of actin filament assembly were monitored using a SHIMADZU RF-5301PC spectrofluorphotometer.
Determination of SRE activity
SRE reporter assays were carried out essentially as described 55. Briefly, NIH3T3 cells were co-transfected with pTK-Renilla and 5× SRE-Lucreporter plasmid provided by A Alberts 37, 38 together with expression plasmids for GFP.DIAPH3 fusion proteins using Lipofectamine 2000. Following transfection, cells were starved in a medium containing 0.5% FCS. 24 h post transfection, cells were lysed and luminescence measurement was performed according to manufacture's instructions (Dual Luciferase Reporter Assay System, Promega).
Statistical analysis and software
Statistical significance was calculated by students t-test analysis (***P < 0.0005; **P< 0.005; *P< 0.05; n.s., not significant). Calculations were performed with Microsoft Excel or GraphPad Prism software, image editing was performed using Adobe Photoshop and Illustrator CS4. For adhesion area measurements, confocal images of cells were acquired at the sites of attachment to the coverglass. Adhesion areas were quantified with the ImageJ software using a freehand tool to define the cell boundary. The ImageJ Stack-Z-function Plugin was applied to generate maximum projection images or sagittal and coronal planes from single z-stacks.
References
Albiges-Rizo C, Destaing O, Fourcade B, Planus E, Block MR . Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J Cell Sci 2009; 122:3037–3049.
Chhabra ES, Higgs HN . The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol 2007; 9:1110–1121.
Charras G, Paluch E . Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 2008; 9:730–736.
Fackler OT, Grosse R . Cell motility through plasma membrane blebbing. J Cell Biol 2008; 181:879–884.
Campellone KG, Welch MD . A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 2010; 11:237–251.
Rottner K, Hanisch J, Campellone KG . WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol 2010; 20:650–661.
Zuchero JB, Coutts AS, Quinlan ME, Thangue NB, Mullins RD . p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol 2009; 11:451–459.
Renault L, Bugyi B, Carlier MF . Spire and Cordon-bleu: multifunctional regulators of actin dynamics. Trends Cell Biol 2008; 18:494–504.
Chesarone MA, DuPage AG, Goode BL . Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol 2010; 11:62–74.
Faix J, Grosse R . Staying in shape with formins. Dev Cell 2006; 10:693–706.
Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S . Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol 1999; 1:136–143.
Hannemann S, Madrid R, Stastna J, et al. The diaphanous-related formin FHOD1 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J Biol Chem 2008; 283:27891–27903.
Takeya R, Taniguchi K, Narumiya S, Sumimoto H . The mammalian formin FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress fibre formation in endothelial cells. EMBO J 2008; 27:618–628.
Hogue M . The effect of hypotonic and hypertonic solutions on fibroblasts of the embyonic chick heart in vitro. J Exp Med 1919; 30:617–649.
Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF . Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 2001; 3:339–345.
Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J . Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol 2001; 3:346–352.
Tokumitsu T, Maramorosch K . Cytoplasmic protrusions in insect cells during mitosis in vitro. J Cell Biol 1967; 34:677–683.
Wolf K, Mazo I, Leung H, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003; 160:267–277.
Bereiter-Hahn J, Luck M, Miebach T, Stelzer HK, Voth M . Spreading of trypsinized cells: cytoskeletal dynamics and energy requirements. J Cell Sci 1990; 96:171–188.
Charras GT, Hu CK, Coughlin M, Mitchison TJ . Reassembly of contractile actin cortex in cell blebs. J Cell Biol 2006; 175:477–490.
Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ . Non-equilibration of hydrostatic pressure in blebbing cells. Nature 2005; 435:365–369.
Cunningham CC . Actin polymerization and intracellular solvent flow in cell surface blebbing. J Cell Biol 1995; 129:1589–1599.
Cunningham CC, Gorlin JB, Kwiatkowski DJ, et al. Actin-binding protein requirement for cortical stability and efficient locomotion. Science 1992; 255:325–327.
Gadea G, de Toledo M, Anguille C, Roux P . Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol 2007; 178:23–30.
Tournaviti S, Hannemann S, Terjung S, et al. SH4-domain-induced plasma membrane dynamization promotes bleb-associated cell motility. J Cell Sci 2007; 120:3820–3829.
Charras GT, Coughlin M, Mitchison TJ, Mahadevan L . Life and times of a cellular bleb. Biophys J 2008; 94:1836–1853.
Riedl J, Crevenna AH, Kessenbrock K, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods 2008; 5:605–607.
Kitzing TM, Wang Y, Pertz O, Copeland JW, Grosse R . Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 2010; 29:2441–2448.
Schwartz MA, DeSimone DW . Cell adhesion receptors in mechanotransduction. Curr Opin Cell Biol 2008; 20:551–556.
Norman L, Sengupta K, Aranda-Espinoza H . Blebbing dynamics during endothelial cell spreading. Eur J Cell Biol 2010; 90:37–48.
Geiger B, Bershadsky A . Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol 2001; 13:584–592.
Block J, Stradal TE, Hanisch J, et al. Filopodia formation induced by active mDia2/Drf3. J Microsc 2008; 231:506–517.
Yang C, Czech L, Gerboth S, Kojima S, Scita G, Svitkina T . Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol 2007; 5:e317.
Pellegrin S, Mellor H . The Rho family GTPase Rif induces filopodia through mDia2. Curr Biol 2005; 5:129–133.
Bartolini F, Moseley JB, Schmoranzer J, Cassimeris L, Goode BL, Gundersen GG . The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J Cell Biol 2008; 181:523–536.
Xu Y, Moseley JB, Sagot I, et al. Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell 2004; 116:711–723.
Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS . Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell 2000; 5:13–25.
Sun H, Schlondorff JS, Brown EJ, Higgs HN, Pollak MR . Rho activation of mDia formins is modulated by an interaction with inverted formin 2 (INF2). Proc Natl Acad Sci USA 2011; 108:2933–2938.
Ramalingam N, Zhao H, Breitsprecher D, Lappalainen P, Faix J, Schleicher M . Phospholipids regulate localization and activity of mDia1 formin. Eur J Cell Biol 2010; 89:723–732.
Peng J, Wallar BJ, Flanders A, Swiatek PJ, Alberts AS . Disruption of the diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr Biol 2003; 13:534–545.
Kitzing TM, Sahadevan AS, Brandt DT, et al. Positive feedback between Dia1, LARG, and RhoA regulates cell morphology and invasion. Genes Dev 2007; 21:1478–1483.
Eisenmann KM, Harris ES, Kitchen SM, Holman HA, Higgs HN, Alberts AS . Dia-interacting protein modulates formin-mediated actin assembly at the cell cortex. Curr Biol 2007; 17:579–591.
Derivery E, Fink J, Martin D, et al. Free Brick1 is a trimeric precursor in the assembly of a functional wave complex. PLoS One 2008; 3:e2462.
Quinlan ME, Hilgert S, Bedrossian A, Mullins RD, Kerkhoff E . Regulatory interactions between two actin nucleators, Spire and Cappuccino. J Cell Biol 2007; 179:117–128.
Pechlivanis M, Samol A, Kerkhoff E . Identification of a short Spir interaction sequence at the C-terminal end of formin subgroup proteins. J Biol Chem 2009; 284:25324–25333.
Watanabe S, Ando Y, Yasuda S, et al. mDia2 induces the actin scaffold for the contractile ring and stabilizes its position during cytokinesis in NIH 3T3 cells. Mol Biol Cell 2008; 19:2328–2338.
Beli P, Mascheroni D, Xu D, Innocenti M . WAVE and Arp2/3 jointly inhibit filopodium formation by entering into a complex with mDia2. Nat Cell Biol 2008; 10:849–857.
Iskratsch T, Lange S, Dwyer J, Kho AL, dos Remedios C, Ehler E . Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J Cell Biol 2010; 191:1159–1172.
Gasman S, Kalaidzidis Y, Zerial M . RhoD regulates endosome dynamics through diaphanous-related formin and Src tyrosine kinase. Nat Cell Biol 2003; 5:195–204.
Gorelik R, Yang C, Kameswaran V, Dominguez R, Svitkina T . Mechanisms of plasma membrane targeting of formin mDia2 through its amino terminal domains. Mol Biol Cell 2011; 22:189–201.
Binder M, Quinkert D, Bochkarova O, Klein R, Kezmic N, Bartenschlager R, Lohmann V . Identification of determinants involved in initiation of hepatitis C virus RNA synthesis by using intergenotypic replicase chimeras. J Virol 2007; 81:5270–5283.
Brown T, Mackey K, Du T . Analysis of RNA by northern and slot blot hybridization. Curr Protoc Mol Biol 2004; Chapter 4:Unit 4 9.
Li F, Higgs HN . Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J Biol Chem 2005; 280:6986–6992.
Brandt DT, Marion S, Griffiths G, Watanabe T, Kaibuchi K, Grosse R . Dia1 and IQGAP1 interact in cell migration and phagocytic cup formation. J Cell Biol 2007; 178:193–200.
Gasteier JE, Madrid R, Krautkramer E, et al. Activation of the Rac-binding partner FHOD1 induces actin stress fibers via a ROCK-dependent mechanism. J Biol Chem 2003; 278:38902–38912.
Acknowledgements
We are grateful for the kind gift of reagents to Art Alberts, Jan Faix and Roland Wedlich-Söldner, to Melanie Schmitt for help with northern blotting, to the SFB638 FACS sorting facility at the ZMBH Heidelberg and to the Nikon Imaging Center at the University of Heidelberg. This research was supported by grants from the Deutsche Forschungsgemeinschaft (GRK 1188 to JS and SK, TRR 17, project A7 to RG), LOEWE (“Tumor and inflammation” to HW) and the Deutsche Krebshilfe (109550 to OTF and RG). OTF is a member of the Cellnetworks cluster of excellence (EXC81).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Plasma membrane blebbing during HeLa cell adhesion depends on Rho-ROCK signaling. (PDF 96 kb)
Supplementary information, Movie S1
A Hela cell stably expressing Lifeact.GFP was imaged during adhesion to an uncoated surface. Live cell imaging was performed using a wide field Olympus IX81 microscope. Frames were taken every 5 seconds for a total period of 5 minutes. (AVI 8818 kb)
Supplementary information, Movie S2
A Hela cell stably expressing Lifeact.GFP was imaged during adhesion to an uncoated surface. Live cell imaging was performed with a wide field Olympus IX81 microscope. Frames were taken every 5 seconds for total period of 15 minutes. 2M Cytochalasin D (Cyto D) was added during imaging as indicated. (AVI 12398 kb)
Supplementary information, Movie S3
A Hela cell stably expressing Lifeact.GFP was imaged during adhesion to an uncoated surface. Live cell imaging was performed with a wide field Olympus IX81 microscope. Frames were taken every 10 seconds for total period of 15 minutes. 10M Y-27632 was added during imaging as indicated. (AVI 12536 kb)
Supplementary information, Movie S4
A Hela cell stably expressing Lifeact.GFP was imaged during adhesion to an uncoated surface. Live cell imaging was performed with a wide field Olympus IX81 microscope. Frames were taken every 10 seconds for total period of 30 minutes. 100M Nocodazole (Noco) was added during imaging as indicated. (AVI 12398 kb)
Supplementary information, Figure S2
Summary of RNAi screen for human actin nucleators involved in blebbing during HeLa cell adhesion. (PDF 112 kb)
Supplementary information, Movie S5
Hela cells stably expressing Lifeact.GFP were imaged during adhesion to an uncoated surface 72 hrs post transfection with control siRNA. Live cell imaging was performed with a wide field Zeiss Axiovert 200M microscope equipped with incubation chamber. Frames were taken every 5 seconds for total period of 5 minutes. (AVI 8404 kb)
Supplementary information, Movie S6
Hela cells stably expressing Lifeact.GFP were imaged during adhesion to an uncoated surface 72 hrs post transfection with DIAPH3 SMARTpool siRNA. Live cell imaging was performed with a wide field Zeiss Axiovert 200 M microscope equipped with incubation chamber. Frames were taken every 5 seconds for total period of 5 minutes. (AVI 8404 kb)
Supplementary information, Figure S3
Distribution of focal adhesions differs between blebbing and non-blebbing cells. (PDF 105 kb)
Supplementary information, Figure S4
Schematic representation of DIAPH3's genomic structure. (PDF 69 kb)
Supplementary information, Figure S5
DIAPH3 variants and their dimerization/actin nucleation deficient mutants during HeLa cell adhesion. (PDF 457 kb)
Supplementary information, Figure S6
DIAPH3 deletion mutants during HeLa cell adhesion. (PDF 473 kb)
Supplementary information, Figure S7
Summary of actin nucleators' GENE IDs and catalogue numbers of SMARTpool siRNAs. (PDF 89 kb)
Supplementary information, Figure S8
DIAPH3 SMARTpool siRNAs sequences and targeting sites. (PDF 49 kb)
Supplementary information, Figure S9
List of qPCR primers used for quantification of actin nucleators' mRNA. (PDF 131 kb)
Rights and permissions
About this article
Cite this article
Stastna, J., Pan, X., Wang, H. et al. Differing and isoform-specific roles for the formin DIAPH3 in plasma membrane blebbing and filopodia formation. Cell Res 22, 728–745 (2012). https://doi.org/10.1038/cr.2011.202
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.202
Keywords
This article is cited by
-
A genome-wide screen for human salicylic acid (SA)-binding proteins reveals targets through which SA may influence development of various diseases
Scientific Reports (2019)
-
A resilient formin-derived cortical actin meshwork in the rear drives actomyosin-based motility in 2D confinement
Nature Communications (2015)
