
Abstract
Interleukin-4 is a cytokine produced by activated T cells, mast cells, and basophils that elicits many important biological responses1 (see Tab 1). These responses range from the regulation of helper T cell differentiation2 and the production of IgE3 to the regulation of the adhesive properties of endothelial cells via VCAM-14. In keeping with these diverse biological effects, high-affinity binding sites for IL-4 (Kd 20 to 300 pM) have been detected on many hematopoietic and non-hematopoietic cell types at levels ranging from 50 to 5000 sites per cell5. This review will focus on the discrete signal transduction pathways activated by the IL-4 receptor and the coordination of these individual pathways in the regulation of a final biological outcome.
Similar content being viewed by others

The cell surface IL-4 receptor complex
The structure of the IL-4 receptor complex was first delineated using chemical cross-linking of 125I-IL-4 to molecules on the surface of IL-4 responsive cells6, 7.
These studies generally found two major complexes crosslinked to 125I-IL-4.One complex contained 125I-IL-4 bound to a protein of˜ 140 kDa. The second complex often contained 80 and 70 kDa molecules cross-linked to 125I-IL-4. These molecules have since been cloned and sequenced.
The molecular cloning of cDNA encoding IL-4 binding proteins8, 9, 10, 11 indicated that s single chain of ˜140,000 Da constituted a high affinity IL-4 binding site (IL-4Rα]. The IL-4Rα bears two pairs of cysteine residues and the WSXWS motif characteristic of the hematopoietin receptor superfamily12, a classic transmenbrane domain, and a very large cytoplasmic domain. A second naturally occuring form of mRNA was observed in murine cells8. This form gives rise to a soluble high affinity IL-4 binding protein of 40 kDa.The nucleotide sequence of the cDNA encoding these molecules indicated that both share the extracellular, ligand binding domain, but that the 40 kDa form lacks the transmembrane and cytoplasmic domains. Examination of the structure of the murine gene13 revealed that exon 8 codes for a soluble form of the IL-4Rα. Thus the two species of receptors are likely to arise from alternative RNA splicing.
The IL-4 receptor complex also contains other polypeptide chains, consistent with the early cross-linking studies. The γ chain of the IL-2 receptor (common γ or γ c) associates with the IL-4Rα chain following the binding of IL-414. This complex has been called the Type I IL-4 receptor15. In addition, the low affinity binding chain for IL-13 (IL-13R-α), a cytokine that elicits biological responses similar to IL-4, can also function as an IL-4Rα associating chain16, 17, 18. The IL-4 receptor containing the IL-13Rα has been termed Type II15.
Both types of IL-4 receptors are competent to signal the activation of similar biochemical signaling events (see below). However, experiments using human B cells have suggested the possibility that there may be some differences in the outcome of signaling via the Type I or Type II IL-4 receptor15. For example, human B cells respond to both IL-4 and IL-13 with a rapid but transient production of inositol (1, 4, 5)-triphosphate followed by an elevation of cAMP19. This has not been observed for murine B cells, which lack expression of the IL-13Rα. The differential response suggests that the IP3/cAMP response may be a result of signaling via the Type II receptor rather than the Type I receptor. More direct experimentation will be needed to clarify the basis for potentially important differences.
Signal transduction by the IL-4 receptor complex
Janus kinases
It has been known for some years that treatment of cells with IL-4 activates cytoplasmic tyrosine kinases20. The Janus family of tyrosine kinases (JAKs) are of critical importance in inititating these pathways21. JAK1 is associated with IL4R-α while JAK3 is coupled to the γ c22, 23, 24. Both JAK1 and JAK3 become tyrosine phosphorylated upon treatment of hematopoietic cells with IL-425, 26. The identity of the kinase(s) associated with IL-13Rα is not yet clear. However, it has been reported that JAK2 or TYK2 can also be tyrosine phosphorylated in response to IL-4 in some cell types27, 28. Heterodimerization of the receptor subunits is thought to activate the kinases and initiate the signaling cascade.
It has been shown that IL-4 can signal in cells expressing the IL-4Rα and either γ c or the IL-13Rα29, 30, 31 demonstrating that the simultaneous expression of both associating chains may not be necessary for signal transduction. Indeed, it was recently observed that IL-4 treatment is able to induce the tyrosine phosphorylation of a STAT (STAT6) in cell lines derived from SCID patients lacking the γ c or JAK3, although less efficently than in normal cells32, 33. These results indicate that in the absence of γ c or in the absence of the kinase to which γ c associates, JAK3, IL-4 signaling can occur, most likely via the Type II IL-4 receptor.
Since both types of IL-4 receptors utilize the IL-4Rα chain, it has been predicted that JAK1 would be critical for signaling IL-4 responses22. Indeed, we and others have observed that the IL-4-induced tyrosine phosphorylation of IRS-1/2 and STAT-6 is impaired in cells lacking the JAK1 kinase34, 35. These results point to a critical role for JAK1 in IL-4 signaling. However, in some cell types tyrosine phosphorylation of JAK1 in response to IL-4 cannot be detected36 suggesting that JAK1 is expressed at very low levels or that some other kinase can replace JAK1 in some circumstances.
Stat6
One of the signaling pathways initiated by IL-4 includes the activation of a latent, cytoplasmic transcription factor that is a member of the signal transducers and activators of transcription (STAT) family37, termed STAT6. The JAK-dependent activation of STATs following engagement of cytokine receptors is associated with their rapid tyrosine phosphorylation and nuclear translocation, presumably allowing these molecules to regulate transcription of cytokine-responsive genes21, 37. As noted above, STAT6 is tyrosine phosphorylated by IL-4 and binds to consensus sequences found within the promoter regions of IL-4-inducible genes38, 39, 40, 41, 42. The JAKl-dependent tyrosine phosphorylation of STAT6 results in its dimerization and translocation to the nucleus. By virtue of its ability to bind to DNA sequences found in the promoter elements of IL-4-responsive genes, STAT6 participates in the IL-4-induced regulation of gene expression.
The insulin receptor substrate
A second signaling pathway activated via the IL-4 receptor involves the insulin receptor substrate (IRS) family (IRS-1 and IRS-2)43, 44. These proteins are large cytoplasmic docking proteins (170-180 kDa) which contain many sites for serine/threonine and tyrosine phosphorylation. Tyrosine phosphorylated sites within IRS-1 and IRS-2 associate with high affinity to cellular proteins that contain SH2 domains, including the p85 subunit of PI-3K, growth factor receptor-bound protein 2 (GRB2), the SH2 and SH3 domain containing adaptor protein nck, the src-family kinase fyn, and the src-homology protein tyrosine phosphatase 2.
Studies examining the functions of IRS1 and IRS2 have indicated that they can signal similarly44. Both are targets for tyrosine phosphorylation induced by treatment of cells with insulin, IGF-I, or IL-4. Both can interact with other signaling proteins as described above. In addition, in IRS1 knockout mice many insulin functions are intact45, 46 suggesting that IRS1 and IRS2 are functionally redundant for insulin signaling. However, a recent study found that there are some differences in the spectrum of proteins recruited to IRS1 as compared to IRS2 and differences in insulin vs IL-4 stimulated cells47. The mechanism of these differences is not clear. Most cells of hematopoietic origin express IRS2, but not IRS1, whereas other cell types may express either one or both43, 44, 48, 49. Human thymocytes and peripheral T cells have been shown to express both50, 51. It is possible that the activation of IRS1 or IRS2 by IL-4 may have subtle consequences for downstream signaling pathways.
Other singaling molecules
IL-4 activates several other singaling molecules including c-fes52 and Shc53 The importance of c-fes in IL-4 singaling is not clear, but some evidence suggests that it may regulate growth and production of IgE in response to IL-454, 55. The importance of Shc in IL-4 signaling is controversial. It has been reported that IL-4 does not induce the tyrosine phosphorylation of Shc28. However, IL-4 clearly induces the tyrosine phosphorylation of Shc in murine B cells53. In addition, a cell line which is highly responsive to IL-4 and lacks IRS expression demonstrates potent Shc phosphorylation in response to IL-4. An additional protein, FRIP-1, that binds to the same region of the IL-4Rα that binds IRS-1/2, is tyrosine phosphorylated in response to IL-456. The roles of these molecules in IL-4 singaling will require further examination.
Regulation of growth
Over the last 5 years numerous studies have examined the mechanism of IL-4 mediated proliferation of cells. Many of these have taken advantage of factor dependent cell lines. The myeloid progenitor cell line 32D expresses endogenous murine IL-4 and insulin receptors but does not express mRNA for either IRS1 or IRS2 and fails to grow in the presence of IL-4, insulin, or IGF-I44, 57, 58. Stable transfectants of 32D expressing IRS1 or IRS2 display both tyrosine phosphorylation of IRS-l/2 and a proliferative response to murine IL-4 treatment as well as to insulin and IGF-I. Transfection of truncation mutants of the human IL4Rα into 32D/IRS1 demonstrated that the region between amino acids 437 and 557 is important for human IL-4 induced IRS-1 phosphorylation and growth in these cells59. This interval contains a single Y residue surrounded by a sequence motif (491PLVIAGNPAYRSFSD505) that is homologous to sequences found in the insulin and IGF-1 receptors. This motif has been designated the insulin-IL-4 receptor motif (I4R motif). Mutation of the central Y residue of the I4R-motif to an F in the hulL-4Rα impairs its ability to signal IRS1 phosphorylation or to induce a proliferative response upon treatment with IL-4 in the majority of expressing clones59. In addition, transfer of a region of the hulL-4Rα containing the I4R-motif to a growth-impaired truncation mutant of IL-2Rβ conveys both IRS1 phosphorylation and sustained proliferation in response to IL-260.
It was proposed a number of years ago that the I4R-motif serves as an IRS1 docking site43. Indeed, the I4R-motif contains an NPXY sequence which is the core sequence thought to interact with phosphotyrosine binding (PTB) domains. A direct interaction between IRS1 and the I4R-motif of the hulL-4Rα was demonstrated using the yeast two hybrid system56, by direct binding of phosphopeptides61, and by solution structure analyses62. These studies revealed that the NPXY region participates directly in binding and that the phosphotyrosine(pY) and hydrophobic amino acids upstream of the pY are critical for interacting with the PTB domain of IRS1. Taken together these results demonstrate that the Y residue in the I4R-motif is important for linking to IRS1 and that the IRS pathway plays a major role in IL-4-mediated proliferation.
Interestingly, this same tyrosine residue can also participate in the binding of the PTB domain of Shc(56), a protein shown to participate in the activation of ras and the regulation of cellular proliferation in other systems. The role of Shc in the IL-4-mediated proliferative response is not understood. Of note is the observation that CT.4S cells, T cells that are IL-4-dependent, do not show the tyrosine phosphorylation of IRS in response to IL-4, but they do show Shc activation. It is possible that depending on the cell type and state of activation either IRS or Shc (or both) signal proliferation.
It was shown that a human IL-4Rα construct terminating at 557 was fully competent to signal proliferation(59), but was greatly impaired at stimultating gene induction and STAT6 gel shift activity (see below). These in vitro studies using receptor mutants suggested that the IRS pathway was critical for growth but that the STAT6 pathway was dispensible. However, three independent laboratories showed that STAT6 knockout mice have a modest to substantial diminution in IL-4-induced co-stimulation of B and T cell growth suggesting that STAT6 is involved in the proliferative response63, 64, 65. The mechanism by which STAT6 might regulate proliferation is not known. When it was cloned, STAT6 was initially thought to be uniquely activated by the IL-4/IL-13 receptors. However, it has recently been shown that anti-IgM or anti- CD40 treatment of splenic B cells66, 67, leptin treatment68, or PDGF treatment69 of responsive cells can also induce the tyrosine phosphorylation of STAT6. It is possible that the defect in lymphocyte proliferation observed in STAT6 deficient animals is due to a defect in some other signaling pathway making the role of STAT6 in IL-4-induced growth an indirect one.
Regulation of gene expression
As discussed above, IL-4 induces the tyrosine phosphorylation of STAT6 which binds to the GAS target sequences found in the promoters of genes such as CD23 and the germline transcript for the heavy chain of IgE, Gɛ. Recent studies of mice with a targeted disruption of the STAT-6 gene clearly demonstrate that STAT6 is important for the induction of genes (CD23, MHC II, Gɛ and IL-4Rα) in response to IL-463, 64, 65. In addition, the ability of the huIL-4Rα constructs to initiate gene expression and STAT6 activation has been analyzed in vitro. A construct terminating at amino acid 657 was fully capable of stimulating gene expression and STAT6 gel shift activity70, while a construct terminating at 557 was greatly impaired at stimultating gene induction and STAT6 gel shift activity. The deletion 557 was not able to signal the tyrosine phosphorylation of STAT6 in response to huIL-4 as detected by western blotting42. These results indicate that the interval between amino acids 557 and 657 is important for signaling the induction of genes and full activation of STAT6 in response to IL-4. This region contains 3 tyrosine residues, falling in a general consensus sequence of GYK/QXF, any one of which can serve to activate the gene induction pathway maximally. This region seems to function independently of the I4R-motif in these transfected cell lines. Transfer of this region of the huIL-4Rα (containing amino acids 557-657) to a truncated IL-2Rβ conveys both STAT6 activation as detected by gel shift assay and potent CD23 induction in response to IL-2, but no growth promotion or IRS phosphorylation. These results suggested that growth and gene induction in response to IL-4 are independently controlled by distinct regions of IL-4Rα. However, recent studies indicate that targets downstream of the IRS pathway actually participate with STAT6 in the regulation of Gɛ induction. It was shown that IL-4 induces the serine phosphorylation of the small, non-histone chromosomal protein HMG(I)Y in an IRS dependent manner71. This phosphorylation was sensitive to wortmannin and rapamycin, inhibitors of PI-3K and the target of rapamycin (TOR) respectively, indicating a role for PI-3K and p70S6K in this process. Previous work from Boothby and colleagues had established that the IL-4-induced serine phosphorylation of HMG(I)Y results in the de-repression of the Gɛ promoter thereby enhancing transcription72, 73.
Regulation of cell survival
IL-4 has profound effects on B and T lymphocytes1. As an important growth and survival factor for normal lymphocytes, IL-4 maintains the viability of small, resting cells, but is unable to induce cellular proliferation without a co-stimulatory agent. Additionally, IL-4 has been shown to reverse the effects of certain stimuli that inhibit B cell activation such as tolerogenic doses of anti-IgM74, 75, the cross-linking of surface IgM and Fc receptors76, and the susceptibility of CD40L-activated B cells to FasL-mediated death77.
In addition, it has been shown that overexpression of IRS1 protects 32D cells from death induced by IL-3 deprivation48. The IRSl-dependent prevention of apoptosis was associated with the activation of PI-3K since wortmannin and LY294002, two inhibitors of PI-3K, partially inhibited the protection from apoptosis mediated by IL-4. Relatively little is known about the mechanisms that transmit signals beyond PI-3K in lymphocytes. Recent reports have placed PKB/Akt, p70 S6 kinase, PKCɛ and PKCn downstream of the activation of PI-3K78. Although it has been demonstrated in several cell types that PI-3K activation leads to the activation of p70S6k79, we found that rapamycin does not block the protection from apoptosis by IL-4 in splenic B cells48, suggesting that p70S6K is not leading to antiapoptotic signals in this system. It is possible that the PI3K pathway leading to the protection from apoptosis in lymphocytes is linked to one of the other downstream effector molecules such as PKB. It has recently been shown that PKB activation by growth factors induces the serine phosphorylation of Bad thereby diminishing its pro-apop-totic activity80. It remains to be determined whether IL-4 treatment of cells also induces such a posttranslational modification.
A second pathway independent of IRS was also found to contribute to the IL-4 induced protection from apoptosis48. Interestingly, this pathway was dependent on the I4R motif of the IL-4Rα since the mutation of Y to F completely abrogated the ability of IL-4 to signal protection from apoptosis. However, this pathway does not include STAT6 since IL-4 is able to protect B and T cells isolated from STAT6 deficient animals from apoptosis as well as in lymphocytes derived from normal mice81, 82. Both Shc and FRIP-1 have been shown to bind the I4Rmotif, and therefore, are good candidates to mediate protection from apoptosis56 although the role they play in IL-4 signaling is currently unclear and will require further efforts for its elucidation.
Both Bcl-2 and caspase families are well know to mediate pro-and anti-apoptotic effects. However, the mechanism by which IL-4 might regulate these proteins is not known. Although there have been reports demonstrating the IL-4-induced regulation of Bcl-2 and Bcl-xL expression in factor dependent lines83, 84, there have been an equal number of reports showing that IL-4 does not regulate their level of expression in regular culture medium85, 86. It is possible that IL-4 regulates these proteins by posttranslational modification as discussed above. Very few studies have addressed the effect of IL-4 on caspase activity. A recent report shows that IL-4 could suppress caspase activity in BCLL cells87; the mechanism used by IL-4 in this process remains undefined, and it will be of interest to see whether the regulation of caspases is a universal mechanism by which IL-4 protects cells from undergoing apoptosis.
Summary
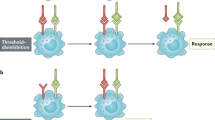
While the early biochemical pathways activated by the binding of IL-4 to its receptor are distinct and independent, it appears that they can overlap further downstream leading to participation in a final biologic outcome (Fig 1). Targets of the IRS pathway, such as HMG(I)Y phosphorylation, act in concert with STAT6 to regulate G-ɛ induction. Targets of the STAT6 pathway act to participate, along with the IRS pathway, in the proliferation response of lymphocytes. Recent data suggests that an unidentified pathway (that is independent of STAT6) acts with the IRS pathway to protect cells from apoptosis. This signaling picture is much more detailed than that put forth in 1994; it is likely that within the next 5 years it will be even more complex.

Signaling pathways emerging from the IL-4 receptor complex. The binding of IL-4 to the IL-4Rαi nduces its association with γ c (or with the IL-13Rα). The Janus family tyrosine kinases JAK1 and JAK3 bind to the IL-4Rα and the γ c cytoplasmic domains, and are thought to become activated when they are brought in close proximity. Activation of the kinases leads to the tyrosine phosphorylation of the IL-4Rα and several signaling molecules including STAT6, IRS, and Shc. The phosphorylation of STAT6 results in its dimerization and translocation to the nucleus where it can bind to promoter elements and participate in the regulation of gene expression. The phosphorylation of IRS results in its association with and activation of other signaling molecules. IRS has been shown to be important in regulating cell survival and entry into cell cycle in response to IL-4. In addition, it is part of a pathway that leads to the serine phosphorylation of the DNA binding protein HMG (I)Y. The function of phosphorylated Shc is not known.
References
Paul WE . Interleukin-4: a prototypic immunoregulatory lymphokine. Blood 1991; 77:1859–65.
Seder PA, Paul WE . Acquisition of lymphokine-producing phenotype by CD4+ T cells. Ann Rev Immunol 1994; 12:635–73.
Coffman RL, Ohara J, Bond MW, Carty J, Zlotnick A, Paul WE . B cell stimulatory factor- 1 enhances the IgE response of LPS-activated B cells. J Immunol 1986; 136:4538–41.
Schleimer RP, Sterbinsky SA, Kaiser J, et al. IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium. Association with expression of VCAM-1. J Immunol 1992; 198:1086–92.
Lowenthal JW, Castle BE, Christiansen J, et al. Expression of high affinity receptors for murine IL-4 on hemopoietic and nonhemopoietic cells. J Immunol 1988; 140:456–64.
Ohara J, Paul WE . Receptors for B-cell stimulatory factor-1 expressed on cells of haematopoietic lineage. Nature 1987; 325:537–40.
Park LS, Friend D, Grabstein K, Urdal DL . Characterization of the highaffinity cell- surface receptor for murine B-cell-stimulating factor 1. Proc Natl Acad Sci USA 1987; 84:1669–73.
Mosley B, Beckmann MP, March CJ, et al. The murine IL-4 receptor: Molecular cloning and characterization of secreted and membrane bound forms. Cell 1989; 59:335–48.
Harada N, Castle BE, Gorman DM, et al. Expression cloning of a cDNA encoding the murine IL-4 receptor based on ligand binding. Proc Natl Acad Sci USA 1990; 87:857–61.
Idzerda RL, March CJ, Mosley B, et al. Human IL-4 receptor confers biological responsiveness and defines a novel receptor superfamily. J Exp Med 1990; 171:861–73.
Galizzi JP, Zuber CE, Harada N, et al. Molecular cloning of a cDNA encoding the human IL-4 receptor. Intl Immunol 1990; 2:669–75.
Cosman D . The hematopoietin receptor superfamily. Cytokin 1993; 5:95–106.
Wrighton N, Campbell LA, Harada N, Miyajima A, Lee F . The murine interleukin4- receptor gene: genomic structure, expression and potential for alternative splicing. Growth Factors. 1992; 6:103–118.
Russell SM, Keegan AD, Harada N, et al. The interleukin-2 receptor chain is a functional component of the interleukin-4 receptor. Science 1993; 262:1880–3.
Callard RE, Matthews DJ, Hibbert L . IL-4 and IL13 receptors: are they one and the same? Immunol. Today 1996; 17:108–10.
Hilton DJ, Zhang J-G, Metcalf D, Alexander WS, Nicola NA, Willson TA . Cloning and characterization of a binding subunit of the IL-13 receptor tht is also a component of the IL-4 receptor. Proc Natl Acad Sci USA 1996; 93:497–501.
Aman MJ, Tayebi N, Obiri NI, Puri RK, Modi WS, Leonard WJ . cDNA cloning and characterization of the human interleukin 13 receptor alpha chain. J Biol Chem 1996; 27146:29265–70.
Caput D, Laurent P, Kaghad M, Lelias J-M, Lefort S, Vito N, Ferrara P . Cloning and characterization of a specific IL-13 binding protein structurally related to the IL-5 receptor alpha chain. J Biol Chem 1996; 271:16921–6.
Finney M, Guy GR, Michell RH . Interleukin 4 activates human B lymphocytes via transient inositol lipid hydrolysis and delayed cyclic adenosine monophosphate generation 1990; 20:151–6.
Keegan AD, Nelms K, Wang LM, Pierce JH, Paul WE . Interleukin 4 receptor: Signaling mechanisms. Immunol Today 1994; 15:423–32.
Ihle JN . Cytokine receptor signaling. Nature 1995; 377:591–4.
Yin T, Tsang ML-S, Yang Y-C . JAK1 kinase forms complexes with IL-4 receptor and 4PS/IRS1 protein and is activated by IL-4 and IL-9 in T lymphocytes. Biol Chem 1994; 269:26614–7.
Miyazaki T, Kawahara A, Fujii H, et al. Functional activation of JAK1 and JAK3 by selective association with the IL-2 receptor subunits. Science 1994; 266:1045–7.
Russell SM, Johnston JA, Noguchi M, et al. Interactions of the IL-2 receptor chains with JAK1 and JAK3: Implications for XSCID and XCID. Science 1994; 266:1042–5.
Witthuhn BA, Silvennoinen O, Miura O, et al. Involvement of the JAK3 kinase in signaling by IL-2 and IL-4 in lymphoid and myeloid cells. Nature 1994; 370:153–7.
Johnston JA, Kawamura M, Kirken RA, et al. Phosphorylation and activation of the JAK3 kinase in response to IL-2. Nature 1994; 370:151–7.
Murata T, Noguchi PD, Puri RK . Receptors for IL-4 do not associate with the common gamma chain, and IL-4 induces the phosphorylation of JAK2 tyrosine kinase in human colon carcinoma cells. J Biol Chem 1996; 270:30829–36.
Welham MJ, Learmouth L, Bone H, Schrader JW . IL-13 signal transduction in lymphohemopoietic cells: Similarities and differences in signal transduction with IL-4 and insulin. J Biol Chem 1995; 270:12286–96.
Lin J-X, Migone TS, Tsang M, et al. Towards a molecular understanding of cytokine peiotrophy and redendancy: IL-2, IL-4, IL-7, and IL-13 activated STAT proteins. Immunity 1995; 2:331–339.
He YW, Adkins B, Furse RK, and Malek TR . Expression and function of theγc subunit of the IL-3, IL-4, and IL-7 receptors. Distinct interactions ofγc in the IL-4 receptor. J Immunol 1995; 154:1596–1605.
Matthews DJ, Clark PA, Herbert J, et al. Function of the IL-2 receptor gamma- chain in biologic responses of X- linked severe combined immunodeficient B cells to IL-2, IL-4, IL-13, and IL-15. 1995; 85:38–42.
Lai SY, Molden J, Liu KD, Puck JM, White MF, Goldsmith MA . IL-4-specific signal transduction events are driven by homotypeic interactions of the IL-4 receptor subunit. EMBO J 1996; 15:4506–14.
Oakes S, Johnston J, Candotti F, et al. Signaling via IL-2 and IL-4 in JAK-3-deficient severe combined immunodeficiency lymphocytes: JAK3-dependent and -independent pathways. Immunity 1996; 5:605–15.
Wang HY, Zamorano J, Yoerkie JL, Paul WE, Keegan AD . The IL-4-induced tyrosine phosphorylation of the insulin receptor substrate (IRS) is dependent on JAK-1 expression in human fibrosarcoma cells. J Immunol 1997; 158:1037–40.
Chen XH, Wang LM, Patel BKR, Flavell RA, LaRochelle WJ, Pierce JH . JAK1 expression is required for mediating tyrosine phosphorylation of signaling molecules in response to IL-4. J Biol Chem 1997; 272:6556–60.
Malabarba MG, Kirken RA, Rui H, Koettnitz K, Kawamura M, O'Shea JJ, Kalthoff FS, Farrar WL . Activation of JAK3, but not JAK1, is critical to interleukin-4 (IL-4) stimulated proliferation and requires a membraneproximal region of IL-4 receptor alpha. J Biol Chem. 1995; 270:9630–7.
Darnell JE, Kerr IM, Stark GR . Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264:1415–21.
Kohler I, Rieber EP . Allergy associated Iepsilon and Fcepsilon receptor II (CD23b) genes activated via binding of an IL-4-induced transcription factor to a novel responsive element. Eur J Immunol 1993; 23:3066–71.
Kotanides H, Reich NC . Requirement of tyrosine phosphorylation for rapid activation of a DNA binding factor by IL-4. Science 1993; 262:1265–7.
Schindler C, Kashleva H, Pernis A, Pine R, Rothman P . STFIL4: a novel IL-4-induced signal transducing factor. EMBO J 1994; 13:1350–6.
Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL . An IL-4 induced transcription factor: IL4 STAT. Science 1994; 265:1701–6.
Quelle FW, Shimoda K, Thierfelder W, et al. Cloning of murine and human Stat6 (IL-4 Stat):A novel stat tyrosine phosphorylated in the responses to IL-4 and IL-3 that is not required for mitogenesis. Mol Cell Biol 1995; 15:3336–43.
White MF and Kahn CR . The insulin signaling system. J Biol Chem 1994; 269:14.
Sun X-J, Wang L-M, Zhang Y, et al. Role of IRS2 in insulin and cytokine signalling. 1995; 377:173–7.
Araki E, Lipes MA, Patti ME, et al. Alternative pathway of insulin signaling in mice with a targeted gene disruption of the IRS1 gene. Nature 1994; 372:186–90.
Tamemoto H, Kadowaki T, Tobe K, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substratel. 1994; 372:182–6.
Sun XJ, Pons S, Wang LM, et al. The IRS2 gene on murine chromosome 8 encodes a unique signaling adapter for insulin and cytokine action. Mol Endocrinol 1997; 11:251–62.
Zamorano J, Wang HY, Wang LM, Pierce JH, Keegan AD . IL-4 protect cells from apoptosis via the insulin receptor substrate pathway and a second independent signaling pathway. J Immunol 1996; 157:4926–34.
Welham MJ, Bone H, Levings M, et al. Insulin receptor substrate-2 is the major 170-kDa protein phosphorylated on tyrosine in response to cytokines in murine lymphohemopoietic cells. J Biol Chem 1997; 272:1377–81.
Johnston JA, Wang LM, Hanson EP, et al. Interleukins 2, 4, 7, and 15 stimulate tyrosine phosphorylation of insulin receptor substrates 1 and 2 in T cells. Potential role of JAK kinases. J Biol Chem 1995; 270:28527–30.
Sharfe N, Roifman CM . Differential association of phosphatidylinositol 3-kinase with insulin receptor substrate (IRS)-1 and IRS2 in human thymocytes in response to IL-7. J Immunol 1997; 159:1107–14.
Izuhara K, Feldman RA, Greer P, et al. Interaction of the cfes protooncogene product with the IL-4 receptor. J Biol Chem 1994; 269:18623–9.
Crowley MT, Harmer SL and De Franco AL . Activation induced association of a 145 kDa tyrosine-phosphorylated protein with shc and syk in B lymphocytes and macrophages. Chem 1996; 271:1145–52.
Izuhara K, Feldman RA, Greer P, Harada N . J IL-4 induces association of the cfes protooncogene product with PI 3 kinase. Blood. 1996; 88:3010–3918.
Schultz C, Izuhara K, Coffman R, Harada N . The critical region in the cytoplasmic domain of human IL-4 receptor for induction of IgE synthesis. Immunol Lett 1995; 46:215–9.
Nelms K, Snow A, Hu-Li J, Paul WE . Identification of a hematopoietic cell specific rasGAP interacting protein. Submitted.
Wang LM, Keegan AD, Li W, et al. Common elements in IL-4 and insulin signaling pathways in factor-dependent hematopoietic cells. Proc Natl Acad Sci 1993; 90:4032–6.
Wang L-M, Myers MG Jr, Sun X-J, Aaronson SA, White M, Pierce JH . Expression of IRS1 restores insulin and IL-4 mediated mitogenesis. 1993; 261:1591–4.
Keegan AD, Nelms K, White MF, Wang LM, Pierce JH, Paul WE . An IL-4 receptor region containing an insulin receptor motif is important for IL-4 mediated IRS1 phosphorylation and proliferation. Cell 1994; 76:811–20.
Wang HY, Paul WE, Keegan AD . IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptoralpha chain. Immunity 1996; 4:113–21.
Wolf G, Trub T, Ottinger E, Groninga L, Lynch A, et al. PTB domains of IRS1 and Shc have distinct but overlapping binding specificities. J Biol Chem 1995; 270:27407–10.
Zhou MM, Huang B., Olejniczak ET, Meadows RP, et al. Structural basis for IL-4 receptor phosphopeptide recognition by the IRS1 PTB domain. Nature Struct Biol 1996; 3:388–93.
Kaplan MH, Schindler U, Smiley ST, Grusby MJ . STAT6 is required for mediating response to IL-4 and for the development of Th2 cells. Immunity 1996; 4:313–9.
Takeda K, Tanaka T, Shi W, et al. Essential role of STAT6 in IL-4 signaling. Nature 1996; 380:627–9.
Shimoda K, Deursen J, Sangster MY, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted STAT6 gene. Nature 1996; 380:630–3.
Karras JG, Wang Z, Coniglio SJ, Frank DA, Rothstein TL . Antigen receptor engagement in B cells induces nuclear expression of STAT5 and STAT6 protein that bind and transactivate an IFN activation site. J Immunol 1996; 157:39–47.
Karras JG, Wang Z, Huo L, Frank DA, Rothstein TL . Induction of STAT protein signaling through the CD40 receptor in B lymphocytes. J Immunol 1997; 159:4350–5.
Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH Skoda RC . Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci USA 1996; 93:6231–5.
Patel BKR, Wang L-M, Lee CC, Taylor WG, Pierce JH, LaRochelle WJ . STAT6 and 6. JAK1 are common elements in PDGF and IL-4 signal transduction pathways in NIH 3T3 fibroblasts. J Biol Chem 1996; 271:22175–82.
Ryan JJ, McReynolds LJ, Wang LH, et al. IL-4-induced growth and gene expression are predominantly controlled by distinct regions of the human IL-4 receptor. Immunity 1996; 4:123–32.
Wang DZ, Zamorano J, Keegan AD, Boothby MR . HMG-I(Y) phosphorylation status as nuclear target regulated through IRS1 and the I4R motif of the IL-4 receptor. J Biol Chem 1997; 272:25083–90.
Kim J, Reeves R, Rothman P, Boothby M . The non- histone chromosomal protein HMG-I(Y) contributes to repression of the immunoglobulin heavy chain germ-line epsilon RNA promoter. Eur J Immunol 1995; 253:798–808.
Wang DZ, Ray P, Boothby M . Interleukin-4-inducible phosphorylation of HMGI(Y) is inhibitedby rapamycin. J Biol Chem 1995; 270:22924–32.
Ales-Martinez JE, Cuenda E, Gaur A, Scott DW . Prevention of B cell clonal deletion and anergy by activated T cells and their lymphokines. Sem in Immunol 1992; 4:195–205.
Parry SL, Hasbold J, Holman M, Klaus GG . Hypercross-linking surface IgM or IgD receptors on mature B cells induces apoptosis that is reversed by costimulation with IL-4 and anti- CD40. J Immunol 1994; 152:2821–9.
Phillips NE, Gravel KA, Tumas K, Parker DC . 1988. IL-4 (B cell stimulatory factor 1) overcomes Fc receptor-mediated inhibition of mouse B lymphocyte proliferation without affecting inhibition of c-myc mRNA induction. J Immunol 1988; 141:4243–9.
Foote LC, Howard RG, Marshak-Rothstein A, Rothstein TL . IL-4 induces Fas resistance in B cells. J Immunol 1996; 157:2749–53.
Franke TF, Kaplan DR, Cantley LC . PI3K: Downstream AKTion blocks apoptosis. Cell 1997; 88:435–7.
Myers MG, Grammar TC, Wang LM, et al. Insulin receptor substrate-1 mediates phosphatidylinositol 3, kinase and p70S6K signaling during insulin, insulinlike growth factor1 and interleukin-4 stimulation. J Biol Chem 1994; 2694:28783–9.
Datta SR, Dudek H, Tao Z, et al. AKT phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91:231–41.
Zamorano J, Austrian J, Wang HY, Keegan AD . Costimulation of resting B lymphocytes alters the IL4 activated IRS2 signaling pathway leading to protection from apoptosis and proliferation: Roles of IRS2 and STAT6. Submitted.
Vella A, Teague TK, Ihle J, Kappler J, Marrack P . IL-4 or IL-7 prevents the death of resting T cells: Stat6 is probably not requried for the effect of IL-4. J Exp Med 1997; 186:325–30.
Danescu M, Rubio-Trujillo, Biron G, et al. IL-4 protects chronic lymphocytic leukemic B cells from death by apoptosis and upregulates bcl-2 expression. J Exp Med 1992; 179:1319–26.
Boise LH, Minn AJ, June CH, Lindsten T, Thompson CB . Growth factors can enhance lymphocyte survival without committing the cell to undergo cell division. Proc Natl Acad Sci USA 1995; 92:5491–5.
Choi MSK, Holman M, Atkins CJ, Klaus GGB . Expression of blcx during mouse B cell differentiation and following activation by various stimuli. Eur J Immunol 1996; 26:676–82.
Minshall C, Arkins S, Straza J, et al. IL4 and insulinlike growth factorI inhibit the decline in Bcl-2 and promote the survival of IL-3-deprived myeloid progenitors. J Immunol 1997; 159:1225–32.
Bellosillo B, Dalmau M, Colomer D and Gil J . Involvement of CED-3/ICE proteases in the apoptosis of B-chronic lymphocytic leukemia cells. Blood 1997; 89:3378–84.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Keegan, A., Zamoran, J. Regulation of gene expression, growth, and cell survival by IL-4: Contribution of multiple signaling pathways. Cell Res 8, 1–13 (1998). https://doi.org/10.1038/cr.1998.1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.1998.1
Keywords
This article is cited by
-
Cytosolic Ca2+ Oscillations in Human Cerebrovascular Endothelial Cells after Subarachnoid Hemorrhage
Journal of Cerebral Blood Flow & Metabolism (2009)
-
Costimulation of resting B lymphocytes alters the IL-4-activated IRS2 signaling pathway in a STAT6 independent manner: implications for cell survival and proliferation
Cell Research (2001)
