
Abstract
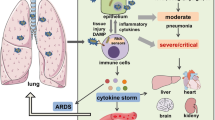
Severe influenza remains unusual in its virulence for humans. Complications or ultimately death arising from these infections are often associated with hyperinduction of proinflammatory cytokine production, which is also known as ‘cytokine storm’. For this disease, it has been proposed that immunomodulatory therapy may improve the outcome, with or without the combination of antiviral agents. Here, we review the current literature on how various effectors of the immune system initiate the cytokine storm and exacerbate pathological damage in hosts. We also review some of the current immunomodulatory strategies for the treatment of cytokine storms in severe influenza, including corticosteroids, peroxisome proliferator-activated receptor agonists, sphingosine-1-phosphate receptor 1 agonists, cyclooxygenase-2 inhibitors, antioxidants, anti-tumour-necrosis factor therapy, intravenous immunoglobulin therapy, statins, arbidol, herbs, and other potential therapeutic strategies.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others


References
Influenza (Seasonal) [World Health Organization, October 2014]. Available from: http://www.who.int/mediacentre/factsheets/fs211/en/.
Taubenberger JK, Morens DM . 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 2006; 12: 15–22.
Knipe DM, Howley PM . Fields Virology. 6th ed. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins Health; 2013.
Liu Q, Liu DY, Yang ZQ . Characteristics of human infection with avian influenza viruses and development of new antiviral agents. Acta Pharmacol Sin 2013; 34: 1257–1269.
Zhang W, Wan J, Qian K, Liu X, Xiao Z, Sun J et al. Clinical characteristics of human infection with a novel avian-origin influenza A(H10N8) virus. Chin Med J 2014; 127: 3238–3242.
Peiris JS, Hui KP, Yen HL . Host response to influenza virus: protection versus immunopathology. Curr Opin Immunol 2010; 22: 475–481.
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG . Into the eye of the cytokine storm. Microbiol Mol Biol Rev 2012; 76: 16–32.
Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011; 146: 980–991.
La Gruta NL, Kedzierska K, Stambas J, Doherty PC . A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 2007; 85: 85–92.
Shinya K, Gao Y, Cilloniz C, Suzuki Y, Fujie M, Deng G et al. Integrated clinical, pathologic, virologic, and transcriptomic analysis of H5N1 influenza virus-induced viral pneumonia in the rhesus macaque. J Virol 2012; 86: 6055–6066.
Hussell T, Goulding J . Structured regulation of inflammation during respiratory viral infection. Lancet Infect Dis 2010; 10: 360–366.
Peiris JS, Cheung CY, Leung CY, Nicholls JM . Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol 2009; 30: 574–584.
Medzhitov R . Recognition of microorganisms and activation of the immune response. Nature 2007; 449: 819–826.
Barton GM . A calculated response: control of inflammation by the innate immune system. J Clin Invest 2008; 118: 413–420.
Wurfel MM . Genetic insights into sepsis: what have we learned and how will it help? Curr Pharm Des 2008; 14: 1900–1911.
Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD et al. Avian influenza A (H5N1) infection in humans. N Eng J Med 2005; 353: 1374–1385.
Ichiyama T, Isumi H, Ozawa H, Matsubara T, Morishima T, Furukawa S . Cerebrospinal fluid and serum levels of cytokines and soluble tumor necrosis factor receptor in influenza virus-associated encephalopathy. Scand J Infect Dis 2003; 35: 59–61.
Muramoto Y, Shoemaker JE, Le MQ, Itoh Y, Tamura D, Sakai-Tagawa Y et al. Disease severity is associated with differential gene expression at the early and late phases of infection in nonhuman primates infected with different H5N1 highly pathogenic avian influenza viruses. J Virol 2014; 88: 8981–8997. English.
Cilloniz C, Pantin-Jackwood MJ, Ni C, Goodman AG, Peng X, Proll SC et al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J Virol 2010; 84: 7613–7624.
de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 2006; 12: 1203–1207.
Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N . Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 2000; 156: 1951–1959.
Jin SQ, Li YY, Pan RG, Zou XF . Characterizing and controlling the inflammatory network during influenza A virus infection. Sci Rep 2014; 4: 1–14. English.
Ramana CV, Cheng GS, Kumar A, Kwon HJ, Enelow RI . Role of alveolar epithelial early growth response-1 (Egr-1) in CD8+ T cell-mediated lung injury. Mol Immunol 2009; 47: 623–631.
Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramirez P et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care 2009; 13: R201.
Brincks EL, Roberts AD, Cookenham T, Sell S, Kohlmeier JE, Blackman MA et al. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J Immunol 2013; 190: 3438–3446.
Darwish I, Mubareka S, Liles WC . Immunomodulatory therapy for severe influenza. Expert Rev Anti Infect Ther 2011; 9: 807–822.
Brun-Buisson C, Richard JC, Mercat A, Thiebaut AC, Brochard L, Group R-SAHNvR. Early corticosteroids in severe influenza A/H1N1 pneumonia and acute respiratory distress syndrome. Am J Respir Crit Care Med 2011; 183: 1200–1206.
Gao HN, Lu HZ, Cao B, Du B, Shang H, Gan JH et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med 2013; 368: 2277–2285.
Xu T, Qiao J, Zhao L, He G, Li K, Wang J et al. Effect of dexamethasone on acute respiratory distress syndrome induced by the H5N1 virus in mice. Eur Respir J 2009; 33: 852–860.
Kudo K, Takasaki J, Manabe T, Uryu H, Yamada R, Kuroda E et al. Systemic corticosteroids and early administration of antiviral agents for pneumonia with acute wheezing due to influenza A(H1N1)pdm09 in Japan. PLoS One 2012; 7: e32280.
Budd A, Alleva L, Alsharifi M, Koskinen A, Smythe V, Mullbacher A et al. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother 2007; 51: 2965–2968.
Zheng BJ, Chan KW, Lin YP, Zhao GY, Chan C, Zhang HJ et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci USA 2008; 105: 8091–8096.
Yao M, Yao D, Yamaguchi M, Chida J, Yao D, Kido H . Bezafibrate upregulates carnitine palmitoyltransferase II expression and promotes mitochondrial energy crisis dissipation in fibroblasts of patients with influenza-associated encephalopathy. Mol Genet Metab 2011; 104: 265–272.
Bassaganya-Riera J, Song R, Roberts PC, Hontecillas R . PPAR-gamma activation as an anti-inflammatory therapy for respiratory virus infections. Viral Immunol 2010; 23: 343–352.
Alleva LM, Cai C, Clark IA . Using complementary and alternative medicines to target the host response during severe influenza. Evid Based Complement Alternat Med 2010; 7: 501–510.
Oldstone MB, Teijaro JR, Walsh KB, Rosen H . Dissecting influenza virus pathogenesis uncovers a novel chemical approach to combat the infection. Virology 2013; 435: 92–101.
Carey MA, Bradbury JA, Rebolloso YD, Graves JP, Zeldin DC, Germolec DR . Pharmacologic inhibition of COX-1 and COX-2 in influenza A viral infection in mice. PLoS One 2010; 5: e11610.
Yuan S . Drugs to cure avian influenza infection – multiple ways to prevent cell death. Cell Death Dis 2013; 4: e835.
Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doerr HW et al. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol 2010; 79: 413–420.
Michaelis M, Geiler J, Naczk P, Sithisarn P, Leutz A, Doerr HW et al. Glycyrrhizin exerts antioxidative effects in H5N1 influenza A virus-infected cells and inhibits virus replication and pro-inflammatory gene expression. PLoS One 2011; 6: e19705.
Ely JT . Ascorbic acid role in containment of the world avian flu pandemic. Exp Biol Med 2007; 232: 847–851.
Palamara AT, Nencioni L, Aquilano K, De Chiara G, Hernandez L, Cozzolino F et al. Inhibition of influenza A virus replication by resveratrol. J Infect Dis 2005; 191: 1719–1729.
Michaelis M, Sithisarn P, Cinatl J, Jr . Effects of flavonoid-induced oxidative stress on anti-H5N1 influenza a virus activity exerted by baicalein and biochanin A. BMC Res Notes 2014; 7: 384.
Kesic MJ, Simmons SO, Bauer R, Jaspers I . Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic Biol Med 2011; 51: 444–453.
Garozzo A, Tempera G, Ungheri D, Timpanaro R, Castro A . N-acetylcysteine synergizes with oseltamivir in protecting mice from lethal influenza infection. Int J Immunopathol Pharmacol 2007; 20: 349–354.
Kurahashi K, Kajikawa O, Sawa T, Ohara M, Gropper MA, Frank DW et al. Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J Clin Invest 1999; 104: 743–750.
Salomon R, Hoffmann E, Webster RG . Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci USA 2007; 104: 12479–12481.
Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol 2007; 81: 2736–2744.
Bayry J, Thirion M, Misra N, Thorenoor N, Delignat S, Lacroix-Desmazes S et al. Mechanisms of action of intravenous immunoglobulin in autoimmune and inflammatory diseases. Neurol Sci 2003; 24: S217–S221
Hung IF, To KK, Lee CK, Lee KL, Yan WW, Chan K et al. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A(H1N1) infection. Chest 2013; 144: 464–473.
Chong PY, Chui P, Ling AE, Franks TJ, Tai DY, Leo YS et al. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: challenges in determining a SARS diagnosis. Arch Pathol Lab Med 2004; 128: 195–204.
Luke TC, Kilbane EM, Jackson JL, Hoffman SL . Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann Intern Med 2006; 145: 599–609.
Fedson DS . Treating influenza with statins and other immunomodulatory agents. Antiviral Res 2013; 99: 417–435.
Liu Z, Guo Z, Wang G, Zhang D, He H, Li G et al. Evaluation of the efficacy and safety of a statin/caffeine combination against H5N1, H3N2 and H1N1 virus infection in BALB/c mice. Eur J Pharm Sci 2009; 38: 215–223.
Viasus D, Pano-Pardo JR, Cordero E, Campins A, Lopez-Medrano F, Villoslada A et al. Effect of immunomodulatory therapies in patients with pandemic influenza A (H1N1) 2009 complicated by pneumonia. J Infect 2011; 62: 193–199.
Nahmod KA, Nahmod VE, Szvalb AD . Potential mechanisms of AT1 receptor blockers on reducing pneumonia-related mortality. Clin Infect Dis 2013; 56: 1193–1194.
Mortensen EM, Nakashima B, Cornell J, Copeland LA, Pugh MJ, Anzueto A et al. Population-based study of statins, angiotensin II receptor blockers, and angiotensin-converting enzyme inhibitors on pneumonia-related outcomes. Clin Infect Dis 2012; 55: 1466–1473.
Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD . CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol 2011; 186: 508–515.
Moseley CE, Webster RG, Aldridge JR . Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir Viruses 2010; 4: 307–311.
Humphreys IR, Walzl G, Edwards L, Rae A, Hill S, Hussell T . A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J Exp Med 2003; 198: 1237–1242.
Kedzierski L, Linossi EM, Kolesnik TB, Day EB, Bird NL, Kile BT et al. Suppressor of cytokine signaling 4 (SOCS4) protects against severe cytokine storm and enhances viral clearance during influenza infection. PLoS Pathog 2014; 10: e1004134.
Zarogoulidis P, Papanas N, Kioumis I, Chatzaki E, Maltezos E, Zarogoulidis K . Macrolides: from in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur J Clin Pharmacol 2012; 68: 479–503.
Blaising J, Polyak SJ, Pecheur EI . Arbidol as a broad-spectrum antiviral: an update. Antiviral Res 2014; 107: 84–94.
Liu Q, Xiong HR, Lu L, Liu YY, Luo F, Hou W et al. Antiviral and anti-inflammatory activity of arbidol hydrochloride in influenza A (H1N1) virus infection. Acta Pharmacol Sin 2013; 34: 1075–1083.
Guidelines for Management of Avian H7N9 Influenza Infection. Available from: http://www.moh.gov.cn/ewebeditor/uploadfile/2013/04/20130410212136993.doc.
Guidelines for Management of Pandemic (H1N1) 2009 Influenza. Available from: http://www.moh.gov.cn/mohwsyjbgs/s9990/200910/43111.shtml.
Chen X, Wu T, Liu G . Chinese medicinal herbs for influenza: a systematic review. J Altern Complement Med 2006; 12: 171–180.
Chen W, Lim CE, Kang HJ, Liu J . Chinese herbal medicines for the treatment of type A H1N1 influenza: a systematic review of randomized controlled trials. PLoS One 2011; 6: e28093.
Poon PM, Wong CK, Fung KP, Fong CY, Wong EL, Lau JT et al. Immunomodulatory effects of a traditional Chinese medicine with potential antiviral activity: a self-control study. Am J Chin Med 2006; 34: 13–21.
Liu Q, Lu L, Hua M, Xu Y, Xiong H, Hou W et al. Jiawei-Yupingfeng-Tang, a Chinese herbal formula, inhibits respiratory viral infections in vitro and in vivo. J Ethnopharmacol 2013; 150: 521–528.
Ling JX, Wei F, Li N, Li JL, Chen LJ, Liu YY et al. Amelioration of influenza virus-induced reactive oxygen species formation by epigallocatechin gallate derived from green tea. Acta Pharmacol Sin 2012; 33: 1533–1541.
Acknowledgements
We acknowledge research funding from the National Nature Science Foundation of China (Grant Nos. 81403163 and 81402404) and Yi Chang Scientific and Technological Bureau (Grant Nos. A14301-04 and A14301-10).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Liu, Q., Zhou, Yh. & Yang, Zq. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 13, 3–10 (2016). https://doi.org/10.1038/cmi.2015.74
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2015.74
Keywords
This article is cited by
-
Interaktion von körperlichen Veränderungen und psychischen Störungen bei COVID-19. Ein Scoping Review
neuropsychiatrie (2024)
-
Immune response in influenza virus infection and modulation of immune injury by viral neuraminidase
Virology Journal (2023)
-
Adjuvant effects of combination monophosphoryl lipid A and poly I:C on antigen-specific immune responses and protective efficacy of influenza vaccines
Scientific Reports (2023)
-
E3 ligase HECTD3 promotes RNA virus replication and virus-induced inflammation via K33-linked polyubiquitination of PKR
Cell Death & Disease (2023)
-
Long COVID and hypertension-related disorders: a report from the Japanese Society of Hypertension Project Team on COVID-19
Hypertension Research (2023)
