
Abstract
As the COVID-19 pandemic is still ongoing, and considering the lack of efficacy of antiviral strategies to this date, and the reactive hyperinflammation leading to tissue lesions and pneumonia, effective treatments targeting the dysregulated immune response are more than ever required. Immunomodulatory and immunosuppressive drugs have been repurposed in severe COVID-19 with contrasting results. The heterogeneity in the timing of treatments administrations could be accountable for these discrepancies. Indeed, many studies included patients at different timepoints of infection, potentially hiding the beneficial effects of a time-adapted intervention. We aim to review the available data on the kinetics of the immune response in beta-coronaviruses infections, from animal models and longitudinal human studies, and propose a four-step model of severe COVID-19 timeline. Then, we discuss the results of the clinical trials of immune interventions with regards to the timing of administration, and finally suggest a time frame in order to delineate the best timepoint for each treatment.
Similar content being viewed by others

Introduction
As evidence of a dysfunctional immune response emerged during SARS-CoV2 symptomatic infection, physicians quickly developed clinical trials to assess the potential effect of immunomodulatory and immunosuppressive drugs in severe COVID-19 patients. However, results from these reports are contrasted. This heterogeneity could be explained by differences in the timing of treatment administration from the onset of infection.
Indeed, both severe COVID-19 clinical and immunological features appear to follow a stereotyped course, similar to other pathogenic coronaviruses (SARS-CoV-1 and MERS-CoV), including several progressive steps from viral inoculation to immune dysregulation leading to tissue injuries. Each of these steps could be considered as a potential therapeutic target.
We postulate that this succession of immune events is mandatory in severe COVID-19 pathogenesis and follows a reproducible timeline, thus helping to determine the best timing for immunomodulation or immunosuppression strategies. Such timing is a key factor in treatment success, as premature immunosuppression could be detrimental by preventing the rise of an efficient immune response while a late therapeutic intervention might not be able to avoid tissue damage and circumvent exhaustion of the immune response.
Our review aims to describe the kinetics of infection and immune response in coronaviruses infection and to reconsider data from immunomodulatory and immunosuppressive drugs clinical studies as well as passive immune interventions with regards to timing, in order to pinpoint the best timepoint for immunomodulation and immunosuppression in COVID-19.
Clinical and biological kinetics of moderate to severe COVID-19: a reproducible course
Despite a certain degree of heterogeneity in the clinical presentation of COVID-19 patients in clinical trials, several clinical and biological features of the disease appear to be consistent between patients and can be considered as milestones of the disease evolution in immunocompetent patients, which might be different in patients with immune deficiencies particularly those with B cell depletion.
To begin with, the incubation period seems to be well-preserved among betacoronaviruses, supporting a reproducible pathogenic course from inoculation to the onset of symptoms. Indeed, a pooled analysis estimated that the incubation period in COVID-19 was 5.1 days (IQR [4.5–5.8]), while it was 5 days (IQR [2–14]) in SARS-CoV and 5–7 days (IQR [2–14]) in MERS-CoV infections1. It should also be noted that the incubation period is relatively homogenous between COVID-19 patients, reflected by the narrow interquartile ranges. Furthermore, in the same study, 97.5% of the patients developed symptoms within 11.5 days (IQR [8.2–15.6]) while only 1% did so 14 days after infection1.
Viral load kinetic also follows a reproducible temporal dynamic, peaking at the day of symptoms onset and then progressively decreasing in three weeks, irrespective of the severity2. Moreover, the timespan of live virus isolation ranges from inoculation to 5–7 days after symptoms onset, as shown by negative viral cultures from pulmonary samples after one week of symptoms and by decreased contagiousness in patients who present symptoms for more than 5 days3,4.
Third, as suggested by a recent modeling study, the course of symptoms follows a stereotyped order, starting with fever, then cough, sore throat, headache, myalgia, nausea and vomiting, and finally diarrhea5.
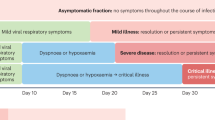
Last, data from observational studies revealed quite early in the pandemic history a reproducible timeline from symptoms onset (DfSO) to disease evolution: dyspnea (5–8 DfSO), hospitalization in general ward (7–11 DfSO), acute respiratory distress syndrome (ARDS) (8–12 DfSO), hospitalization in ICU (10.5–12 DfSO), the need of IMV (invasive mechanical ventilation) (8–10.5 DfSO) or mortality (16–18.5 DfSO) (Table 1). These early epidemiological data were later confirmed by retrospective cohort studies and clinical trials. However, we acknowledge that some factors of heterogeneity in the disease course remain, due for example to the occurrence of severe thrombosis and/or secondary bacterial infections. Thus, despite a high level of reproducibility, these timepoints may not account for all patients trajectories.
Additionally, and although being helpful to evaluate the stage of the disease, the DfSO assessment relies on patient’s declaration and perception of the first symptoms and thus implies variability. To outmatch this limit, other indicators of the disease course can be used as landmarks, such as biological markers of inflammation and oxygen levels requirements. Indeed, many studies reported a significant association between increased CRP, IL-6, IL-10, D-dimers, LDH, calprotectine or ferritin levels and severe forms of the disease6,7. However, only few studies correlated these markers with DfSO, and very few information is available regarding their levels in symptomatic patients before oxygen requirement. After reviewing clinical trials with homogenized population, it appears that CRP level gradually increases with WHO clinical progression scale (Fig. 1). Indeed, WHO score 5 patients presented a median CRP at ~100 mg/L, whereas in patient with WHO scores 6–9, CRP levels were around 150 mg/L7,8. However, the direct correlation between CRP levels and each WHO score of severity has not yet been demonstrated. Oxygen supplementation level is also a promising indicator of COVID-19 course. Indeed, from the RECOVERY preliminary report, patients without oxygen supplementation at inclusion had a median DfSO of 6 days (IQR [3–10]), while patients requiring oxygen supplementation had a median DfSO of 9 days (IQR [5–12]), and those who needed IMV had a median DfS0 of 13 days (IQR [8–18])8.

WHO clinical progression scale (reproduced from www.who.int12).
Overall, several markers can be used to specify the disease progression, such as DfSO, oxygen levels, biological inflammation markers (including neutrophil to lymphocyte ratio) and radiological scores9,10,11. Combining these data could provide an accurate estimation of the disease stage.
According to the aforementioned information, we hereunder suggest a five-step schematic clinical course of severe COVID-19, where each phase could be a target for specific therapeutic interventions: the first phase corresponding to the incubation period (from infection to the day of symptoms onset (DfSO): DfSO – 5 to 0, WHO score 112); a second phase corresponding to the viral phase (from symptom onset to dyspnea: DfSO 0–7, WHO scores 2–4), a third phase corresponding to the state of inflammatory pneumonia (DfSO 7–12, oxygen requirement, WHO score 5, high acute phase reactant levels), a fourth phase corresponding to the brutal clinical aggravation reflected by acute respiratory distress syndrome (ARDS) (DfSO 12–18, high flow oxygen, WHO scores 6–9, high acute phase reactant levels) and finally, in survivors, the fifth phase potentially including lung fibrosis, and/or persisting in the form of “post-covid” symptoms (some cases pertaining to the long-covid status). Of note long-COVID usually follows an infection with a benign course and its physiopathology remains to be elucidated and will not be discussed here.
Timeline of the immune responses in severe COVID-19
Incubation period (DfSO – 5 to 0)
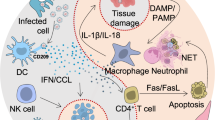
The very first immune events following SARS-CoV-2 infection are described in several animal models, mainly Angiotensin-converting-enzyme (ACE)-induced transgenic mice and non-human primates (NHP). Following inoculation, the virus infects type 1 and 2 pneumocytes through the ACE2 receptor, leading to Nuclear factor kappa B (NF-kB) pathway activation13,14. Local production of chemokines (CXCL-10, CCL-2, CCL-4) and cytokines (IL-6, TNF-α, IL-1RA, IFN-α, IFN-β) is induced from day 1 post-infection (DPI)15,16,17, as observed in SARS-CoV and MERS-CoV infections featuring chemokine (CCL-10, CCL-2, CCL-3) and cytokine (IFN-α, TNF-α, IL-2, IL-12/23, IL-6) production from 1–3 DPI18,19,20. This first influx drives monocytes, plasmacytoid dendritic cells and lymphocyte attraction to the lung peri-vascular and peri-bronchial spaces from 2–3 DPI15,16, preceding or concomitant with the first symptoms.
Type I IFN have emerged as key early determinants of COVID-19 severity. Unlike other viral infections, SARS-CoV-2 induces little amounts of IFN, primarily type I (α and β) and type III (λ)21,22. Most severe and critical patients exhibit low amounts of circulating type I IFN and a diminished IFN blood signature. Moreover, inherited deficiencies in IFN-I pathways and auto-antibodies against all IFN-I subtypes have been associated with severe forms of COVID-1923,24. Virus escape-strategies from the IFN system have been reported13, through direct inhibition of IFN-I production and signaling, as well as antagonism of IFN-I receptor by infection-induced circulating IgG antibodies25,26. This impairment of the early IFN-I responses could result from direct inhibition of STAT-1 by SARS-CoV-2 NSP1 and ORF6, and subsequent compensatory upregulation of the STAT-3 pathway, leading to coagulopathy in part due to complement activation and cytokine production27,28,29. Interestingly, IFN-III responses have also been reported to be down-tuned and delayed in COVID-19, which could reflect an abnormal immune response from infected epithelial cells at the early stage30.
Second phase: from symptoms onset to dyspnea (DfSO 0–7)
At this stage, the production of pro-inflammatory cytokines increases, likely produced by the immune cells recruited in the lungs31. IL-6, IL-1β, TNF-α levels rise in the lungs, and increase in plasma, accounting for the first symptoms32,33. In mice, the initial transcriptional signature of immune cells in the lungs switches from type I and II IFN signaling, neutrophil recruitment and PRR activation to cytokine signaling, type II IFN and neutrophil recruitment15. Several other cytokines are also overproduced, including IL-8, IL-10, IL-1516,17,34, as well as chemokines (CXCL-10, CCL-2, CCL-3, CCL-5, CXCL-17)15,35, leading to an increase in the immune cell lung infiltration, extending to the alveolar space15,36. This innate immune response allows for viral control with decreasing viral replication2. During the first week following symptoms onset, peripheral B lymphocyte counts increase, mainly represented by double-negative B lymphocytes (lacking IgD and CD27 expression) and plasmablasts in severe patients, reflecting the absence of germinal center maturation and predominance of extrafollicular responses37,38,39. While at this step, specific antibodies titers rise in non-severe COVID-19 patients with detectable specific IgM and IgA from 5–7 DfSO, and specific IgG from 7–10 DfSO37,40,41, severe cases feature delayed antibody production and lower proportion of neutralizing antibodies during the first week of symptoms, but do not differ in terms of antibody levels41,42,43. Interestingly, pre-immunization to human betacoronaviruses could dampen the antibody response to specific SARS-CoV-2 epitopes and theoretically mediate severity41.
Third phase: from dyspnea to ARDS (DfSO 7–12)
Several longitudinal human studies described the immune response kinetics from 6 DfSO and confirmed the global increase in IL-6, TNF-α, IFN-γ, but also IL-10, associated with an NF-κB signature, as well as IL-8, IL-15, while they found discrepancies in the circulating IFN-α2 levels33,44,45. The STATs/IRFs pathways are also activated and amplify the cytokine cascade, as shown for STAT1/IRF346 and STAT347,48,49. The early over-production of IL-10 has been proposed as a specific feature of COVID-19 infection, and could participate in the immune cell dysfunction and systemic inflammation by over-activating CD8 T cells and mast cells50,51. Importantly, the elevated levels of TNF-α and IFN-γ were reported to have a synergistical effect on inflammatory cell death induction in a mice model of COVID-19, and treatment with combined neutralizing antibodies towards both cytokines were able to prevent mortality and cytokine storm52. Furthermore, G-CSF and GM-CSF have been found to be elevated in both ICU and non-ICU patients compared to healthy controls, and correlated with severity of symptoms53. Interestingly, the early cytokine signature (before 12 DfSO) segregated patients with severe outcomes, enlightening the importance of early immune events to predict disease evolution45.
Following the second phase, patients with only moderate disease likely develop functional specific CD4 Th1 and follicular helper T cell (TFh) responses, including effector and central memory subsets, that are able to tune down the inflammation, as demonstrated in patients who were recovering from COVID-19, but also from SARS-CoV and MERS-CoV infections54,55,56,57,58. However, severe patients do not mount an effective and functional T cell response, they produce more inflammatory cytokines and thus evolve towards the severe stage of the disease59. These findings are illustrated by the lower proportion of IFNy+ CD4 T cells observed in severe cases and in patients with comorbidities, further linking impaired Th1 response with disease severity59,60. Moreover, dysregulation of TFh subsets might account for the elevated number of circulating plasmablasts reported in severe forms37,44,61. The failure in developing adequate T cell responses to SARS-CoV-2 might be due to impaired antigen-presentation abilities in dendritic cells, as observed during the first 3 weeks of the infection, including reduced expression of costimulatory molecules CD80/CD86, reduced proliferation and IFNγ / TNF-α production by T cells harboring both HLA-DR and PD-1 simultaneously62. This hypothesis is supported by the observation of HLA class II downregulation on myeloid cells31,63, a feature already described in SARS-CoV-1 mice models64 that was associated in vivo with IL-6, IL-8 and CXCL-10 production65 and that could be potentially due to IL-6 in particular66. Lastly, a decrease in circulating plasmacytoid dendritic cell has been reported, possibly due to recruitment in the lungs as shown in NHP and associated with pro-inflammatory activity17,62.
Fourth phase: ARDS (DfSO 12–18)
Patients with a severe form of Covid-19 develop hyperactivated and dysfunctional T cells, mainly TH1 with evidence of impaired contraction67,68,69, and some with skewed phenotype towards TH2 and/or TH17 differentiation leading to production of IL-4, IL-5, IL-13, IL-17A (a feature also observed in NHP models70 and shared with SARS-CoV-1)37,45,71,72. A sustained inflammation with a rebound in IL-6, IL-8, IL1-β, TNF-α from 10–16 DfSO33 leads to complement activation (with reported elevation of soluble C5a and over-expression of C5aR1 on both circulating and pulmonary myeloid cells)29,73, as well as production of VEGF53, increase in circulating immature neutrophils and myeloid-derived suppressor cells7,31,33 and exhaustion of T cell phenotype44, thus driving diffuse alveolar damage and ARDS. Interestingly, an over-production of pro-inflammatory afucosylated IgG antibodies has also been reported in ARDS patients. It correlates with severity and might contribute to the inflammation loop through increased NK cell degranulation and monocyte cytokine production74,75. The conjunction of myeloid cells activation, complement cascade stimulation, systemic inflammation and viral-induced endotheliitis may also lead to a pro-thrombotic state responsible for significant morbi-mortality76,77,78,79. At this stage, pneumonitis is associated with a neutrophilic and lymphocytic infiltration80. The late evolution of ARDS in survivors features signs of pulmonary fibrosis, whose mechanism remains to be elucidated81.
Overall, these data suggest an early immune response mediated by epithelial cell cytokine and chemokine secretion, followed by an increase in monocytes and inflammatory macrophages responsible for systemic inflammation and notably IL-6 peaks around 6–10 DfSO (Fig. 2). In severe forms, a deficiency in antigen-presentation abilities by dendritic cell might prevent the generation of an appropriate Th1 T cell responses, leading to uncontrolled inflammation and to ARDS. Finally, in ARDS survivors, exhausted lymphocytes and recruitment of myeloid-derived suppressor cell might contribute to a pro-fibrotic environment.

IFN interferons, DfSO days from symptoms onset, ICU intensive care unit, ARDS acute respiratory distress syndrome, NIV non-invasive ventilation, IMV invasive mechanical ventilation, *in patients with impaired type I IFN response.
Proposed timepoints for immune-interventions in COVID-19
First phase or pre-emptive treatment: from inoculation to first symptoms (DfSO−5 to 0, WHO score 1)
As previously stated, the early immune responses to COVID-19 are driven by viral replication, peaking the day of symptom onset or shortly before2. Thus, pre-emptive treatment should provide antiviral effects rather than counter-acting the initial immune response (Fig. 3).

IFN interferons, DfSO days from symptoms onset, ICU intensive care unit, ARDS acute respiratory distress syndrome, NIV non-invasive ventilation, IMV invasive mechanical ventilation, *in patients with impaired type I IFN response.
Although the SOLIDARITY trial did not report a beneficial effect of subcutaneous interferon beta-1), recent phase 2 studies reported positive effects of inhaled interferon beta-1 and subcutaneous peginterferon lambda in the first and second phase of the disease, keeping the door open for interferon-based therapies at these stages82,83,84. Another point to consider is that IFN beta at low dose was used in SOLIDARITY, and that different IFN doses might have distinct antiviral efficacy. Moreover, therapeutic strategies using other type I IFN subtypes, such as alpha 2 or even other alpha-subtypes, as well as type III (lambda) IFN, could induce different responses as observed with other viruses such as hepatitis C virus.
Monoclonal antibodies directed towards the receptor-binding-domain of the Spike protein have shown positive results at this pre-emptive stage. Indeed, the association of Imdevimab and Casirivimab could reduce the risk of symptomatic COVID-19 in non-infected patients by 81% and by 31% in already infected but asymptomatic patients85. However, their high cost production and relatively low availability especially in the pre-hospital setting could restrict their use at a larger scale and overall should be used at early stage only in patients at risk with no antibodies against the virus, preferentially in patients with poor B cell response in primary and secondary immune deficiency.
A recent screening of 1900 clinically safe drugs identified masitinib, a Tyrosine-Kinase inhibitor, as a promising anti-viral therapy. Indeed, masitinib was able to reduce in vitro SARS-CoV-2 replication by inhibiting the SARS-CoV-2 main protease 3CLpro86, and should be further investigated in clinical trials.
Last, theorically, using anti-cytokine drugs such as JAK inhibitors or anti-IL1/IL6 therapies at this stage could impair viral clearance and increase direct viral toxicity. To our knowledge, no clinical trial has evaluated the effect of a pre-emptive immunomodulatory treatment in COVID-19, and therefore should not be used in this stage.
At this step, ~60–80% of patients will evolve toward a symptomatic disease87,88.
Second phase or early treatment: from symptoms onset to dyspnea (DfSO 0–7, no oxygen needed, WHO scores 2–4)
After a median incubation time of 5 days1, the innate immune response triggers a decline in viral replication, circulating monocytes are recruited in the lower airway tract and differentiate into inflammatory macrophages, leading to increased cytokine production and systemic inflammation, inducing inflammatory damage89. Thus, at this stage the aim is more to prevent the dysregulated immune response rather than fighting against the viral replication. Therefore, this disease stage appears to be theoretically the first launch window for immunomodulatory therapies, and possibly the optimal timepoint for therapeutic interventions in order to prevent pneumonitis. However, as most immunomodulatory drugs have been tested in patients already receiving oxygen support, one can only speculate about the benefits of an earlier administration, i.e., before oxygen requirement.
Considering the positive results of anti-IL-6 therapies at the latter stages of the disease, and taking into account the rather long half-life of Tocilizumab (estimated at 10 days90), an earlier administration in selected patients could be discussed. Moreover, as pointed out by the RECOVERY trial, Tocilizumab was able to reduce 28-day mortality in patients treated before 7 DfSO, although almost all included patients were under oxygen support at inclusion91. This strategy should be further investigated.
As discussed above, GM-CSF antagonists have also shown promising results in COVID-19 at a later stage, yet no study to date has described the effect of an earlier treatment. However although such a strategy could reduce myelopoiesis, monocyte recruitment in the lungs and differentiation into inflammatory macrophages, and could be even more beneficial92.
In contrast, dexamethasone seems to be not effective at this stage and could even be deleterious. In the RECOVERY study, the use of dexamethasone in patient without oxygen with a median at 6 DfSO IQR [3–10], was not associated with improved survival (RR 1.19 (95% CI, 0.91–1.55))8. In addition, in a retrospective multicenter study, the use of glucocorticoids in patients with CRP < 100 mg/L was harmful, but was beneficial when CRP was over 200 mg/L93.
Convalescent plasma therapy could be beneficial at the earlier stages, as suggested by animal studies showing a greater effect when given early after inoculation, but this strategy has yet to be tested in humans94. In a small study using propensity-score matched controls, authors reported a positive effect on oxygen levels requirement and survival among 39 patients with severe COVID-19, greater in patients treated before 7 DfSO95. Moreover, a recent randomized controlled trial including 160 elderly patients (mean age 76.4 years old, SD 8.7) showed a reduced risk of progression to severe COVID-19 when administered between 0 and 3 DfSO96. Interestingly, another randomized trial including 333 slightly younger patients (median age: 62 years old (IQR[53–72.5]) reported disappointing results, possibly because of late administration (at a median of 8 DfSO (IQR [5–10]))97. It is likely that the beneficial effect of convalescent plasma therapy relies on its antiviral properties rather than its immunomodulatory effects, and thus should be considered during active viral replication phases, i.e., from inoculation to 7 DfSO, with the exception of immunocompromised patients who display delayed viral clearance as a consequence of late or no immune response (particularly in patients with B cell deficiencies) and thus might retain benefit at later timepoints98.
Anti-Spike monoclonal antibodies could also be useful at this stage: the aforementioned combination of Imdevimab and Casirivimab was reported to reduce the composite risk of hospitalization or death by 70–71% in high-risk non-hospitalized infected patients99, and the association of Bamlanivimab and Etesevimab reduced the combined risk of hospitalization or death among patients with mild to moderate COVID-19 in a large randomized controlled trial (BLAZE-1 study100), yet Bamlanivimab does not seem to retain activity against the actively spreading L452R delta variant101.
Other strategies are currently being investigated at this stage, among which IL-7 agonists in lymphopenic patients (NCT04407689), and anti-IL-4/IL-13 antibodies in hospitalized patients (NCT04920916). Despite several calls for investigation, a strong rationale and promising observational data in patients treated for inflammatory bowel disease, phase I and II studies evaluating the effects of a TNF-alpha antagonist strategy are scarce and a few are ongoing (NCT04425538, NCT04705844)102,103. Anti-PD-1 agents are also investigated in obese COVID-19 patients treated before 7 DfSO, aiming at reducing the associated immune dysfunction and thus improve survival104.
Overall, immunomodulation is a theoretical promising approach at this timepoint and could slow down the evolution towards pneumonitis, but this hypothesis needs to be confirmed. Moreover, initiating immunomodulation at this stage exposes to the risk of over-treatment and thus would require accurate predictive scores to better select high-risk patients who could benefit of such an approach. However, immunosuppressive therapies (i.e., high-dose steroids) does not seem to be effective, and could even be harmful.
At this step, nearly 19% of symptomatic patient will develop dyspnea and require oxygen therapy105.
Third phase treatment: from dyspnea to ARDS (DfSO 7–12, oxygen requirement, WHO score 5, high acute phase reactant levels)
When symptoms evolve and require hospital admission, emergency immunomodulatory therapies could prevent the evolution into an auto-amplifying inflammatory loop leading to ARDS. Dexamethasone was the first therapy that showed reduction of 28-day mortality in patients who were receiving oxygen without ventilatory support and only in patients treated after 7 DfSO (Table 2A)8. In another study, glucocorticoids were effective on mortality only in patients with WHO score 5 (receiving oxygen >3 L/min) and in patients with CRP level >100 mg/L106 (Table 2A). Dexamethasone is now recommended at this stage by the WHO and National Institutes of Health107.
Several trials assessed the effect of IL-6 antagonists at this step, with contrasting results. In the RECOVERY trial, Tocilizumab was not significantly associated with better survival in patients with oxygen only and in patients with non-invasive ventilation (RR 0.84; 95% CI, 0.69–1.03 and RR 0.86; 95% CI, 0.74–1.01, respectively)91. The EMPACTA trial showed a reduction of the composite risk of intubation or death in patients with WHO score 5 treated by Tocilizumab108 and the CORIMUNO-19 study also showed a potentially beneficial effect of Tocilizumab in patients with WHO score 5 at 14 DfSO109. On the other hand, the TOCIBRAS110, COVACTA111, BACC Bay112, COVINTOC113 or RCT-TCZ-COVID-19114 trials did not detect a positive effect of Tocilizumab in WHO score 5 patients, as well as with Sarilumab115. Finally, a recent meta-analysis of prospective randomized clinical trials conducted by the WHO pinpointed a reduction in the all-cause mortality risk at 28 days (OR 0.86 (95% CI, 0.79–0.95; P = 0.003) for all IL-6 antagonists, OR 0.83 (95% CI, 0.74–0.92; P < 0.001) for Tocilizumab, and OR 1.08 (95% CI, 0.86–1.36; P = 0.52) for Sarilumab), but also with a lower risk of progression to IMV, cardiovascular support, and kidney replacement therapy in patients receiving IL-6 antagonists included in randomized controlled trials116. Although no precise data on timing of administration was provided, the OR for 28-day mortality were lower in patients treated while under oxygen support <15 L/min than in patients requiring IMV at treatment initiation, and lower in patients co-treated with corticosteroids. The use of Tocilizumab has consequently been recently incorporated in the WHO Guidelines for Covid-19 management117.
Targeting IL-1 at this stage remains controversial. The preliminary results from the randomized controlled trial ANACONDA suggested a detrimental effect118,119,120 and the randomized clinical trial CORIMMUNO-ANA-1 did not report an improvement in patients treated at a median of 10 DfSO whatever the end point (survival, duration of oxygen requirement, etc.)121. Another IL-1 antagonist, targeting IL-1beta, failed to improve survival at day-29 in hypoxic patients treated before mechanical ventilation122. However, a recent phase III trial reported a protective effect of Anakinra when administered in selected patients with increased soluble urokinase plasminogen activator receptor at a median of 9 DfSO, reducing 28-days mortality (Hazard ratio 0.45), and a meta-analysis including 15 retrospective and prospective studies for a total of 757 patients treated with Anakinra found a protective effect on 28-day mortality (OR 0.34; 95% CI, 0.21–0.54), mainly administered in patients with either strong inflammatory features or severe COVID-19 pneumonia, leaving the door open for IL-1 antagonists at this stage123,124.
JAK-inhibitors have also been evaluated in COVID-19, and showed promising results, possibly counter-acting the strong type 1 IFN signature reported in immune cells lung infiltrates, as well as reducing signaling of other inflammatory cytokines31,125. In a large randomized controlled trial, Baricitinib, a JAK1/2 inhibitor, administered at a median of 8 (IQR [5–10]) DfSO, reduced the time to recovery when associated with Remdesivir and compared to Remdesivir alone, especially in WHO scores 5 and 6 patients126. In another large randomized controlled trial, Tofacitinib, a JAK1/3 inhibitor, significantly reduced the cumulative risk of death or respiratory failure at day-28 (RR 0.63; 95% CI, 0.41–0.97; P = 0.04) in patients with COVID-19 pneumonia not requiring mechanical ventilation at inclusion, at a median of 10 (IQR 7–12) DfSO127 (Table 2C). Overall, these data suggest a beneficial effect of JAK-inhibitors in patients with severe COVID-19 and requiring oxygen support, but before mechanical ventilation and ARDS. However, these JAK inhibitors by blocking JAK1 may impair type 1 IFN signaling required for virus clearance.
Likewise, IFN-I recombinant therapy could be beneficial also at this stage in the subgroup of patients with impaired type 1 IFN response and may prevent the evolution towards ARDS44.
Several studies reported promising results from GM-CSF antagonists in COVID-19, and a recent randomized controlled trial found a 65% reduction in the risk of mechanical ventilation or death at day 29 in non-mechanically ventilated patients with hypoxia and severe COVID pneumonitis treated with Mavrilimumab (an anti-GM-CSF receptor alpha antibody) (HR 0.36, p = 0.0175, press release128). Furthermore, anti-GM-CSF antibodies appear to be effective in patients aged over 70 years old and hospitalized for severe COVID-19, as results from the phase II OSCAR trial (evaluating the drug otilimab) reported a higher probability of being alive and free of respiratory support at day 28, as well as a reduced 60-day mortality129. Anti-VEGF antibodies could also be helpful at this stage by reducing oxygen-support duration in patients with hypoxemic pneumonia through modulation of abnormal vascularization but also by a immune modulatory effect130. While the c-Kit inhibitor Masitinib will be investigated for it’s promising antiviral action, another phase II clinical trial also is currently evaluating its effect in non-ICU hospitalized patients with moderate to severe COVID-19 pneumonia in association with a disulfide isomerase inhibitor, Isoquercetin131. State of the art randomized controlled trials will be warranted to confirm these promising signals.
Overall, at this stage, while glucocorticoids seem to be the main therapeutic option, there might be room for immunomodulation. Moreover, a synergistical effect of Tocilizumab and Dexamethasone has been reported in the RECOVERY trial, (increase survival of the combinational treatment when compared to Tocilizumab alone (RR 0.80; 95% CI, 0.70–0.90, compared to RR 1.16; 95% CI, 0.91–1.48, respectively)91, and is currently investigated in the dedicated TOCIDEX trial, in comparison to Dexamethasone alone132.
After this phase, 5% of patients will require ICU and 2.3% will need IMV105.
Fourth phase: ARDS (DfSO 12–18, high flow oxygen and mechanical invasive ventilation, WHO score 6–9, high acute phase reactant)
At this stage, patients present with a severe pneumonia featuring increased lung infiltration by neutrophils and activated lymphocytes, leading to severe local inflammation and organ damage, causing ARDS80. Patients requires IMV (WHO score 7–9). Thus, the main goal is to suppress the existing inflammatory lung infiltration without being too deleterious regarding late viral clearance and secondary ICU bacterial infections.
To this end, Dexamethasone has shown the most encouraging results. Indeed, in the RECOVERY report, the greater efficacy of Dexamethasone on mortality was observed in intubated patients (RR, 0.64 (95% CI, 0.51–0.81))8. Accordingly, the REMAP-CAP study also suggested a benefit for hydrocortisone in ICU COVID-19 patients133 (Table 2A).
Immunomodulation also appears to be effective at this later stage. Indeed, while in the RECOVERY study Tocilizumab was not associated with better survival at day 28 when used in intubated patients (possibly because of premature timepoint)91, in the REMAP-CAP report, Tocilizumab and Sarilumab improved outcomes including survival at 90 days in critically ill patients. Moreover, a large retrospective study of patients admitted in ICU showed that Tocilizumab was associated with increased survival134, whereas a post-hoc analysis of the COVINTOC trial suggested that Tocilizumab could be effective in patients with score 6 and more113. Finally, in the international double blind randomized clinical trials Sarilumab increased survival by 8.9% in WHO class 6 patients (of whom only 20% also received corticosteroids), but this difference was not statistically significant (p = 0.25)115. Overall, Tocilizumab is now recommended at this stage by the WHO and FDA135,136.
Moreover, the previously discussed anti-JAK1/3 antibody Tofacitinib could also be considered at this stage in patients receiving high-flow oxygen; as the recent randomized controlled trial reported a trend in the reduction of the combined risk of death or respiratory failure in WHO ordinal scale 6 patients (OR 0.62, (95% CI [0.15–1.79]))127.
Complement-mediated inflammation can be induced directly by SARS-COV-2 surface proteins, elevated levels of C5a and an association between sC5b-9 and PO2/FiO2 have been reported in ARDS, making complement-targeted therapies a promising option to reduce inflammation and coagulopathy, with interesting results from early studies, but disappointing results from the ALEXION trial in WHO class 5 and above patients73,137,138,139,140.
Anti-VEGF agents could also be beneficial at this stage through modulation of the abnormal angiogenesis and potential immunomodulatory effects, and should be further evaluated in patients requiring mechanical ventilation.
Last, anti-fibrotic therapies have also been discussed in ARDS patients, although their benefit remains to be proven141,142.
Conclusion & perspectives
While antiviral therapies, because of the lack of highly efficient drugs apart from vaccination, showed disappointing results, immune interventions have proven to be beneficial in COVID-19, but the best drugs and timing for their administration has yet to be determined82. After reviewing basic science studies and clinical trials data, we have shown that COVID-19 infection is characterized by a stereotyped pattern of immunological events, one leading to another, and thus follows a reproducible timeline of immune dysregulation steps, each representing potential targets for immune interventions. Overall, severe COVID-19 course begins with an imbalanced innate immune cells activation, leading to defective antigen presentation and impaired T & B cell responses, altogether contributing to increasing and unrestrained systemic inflammation and ARDS. To this date, immune interventions have shown beneficial effects in patients with WHO score 5 and forward disease. Considering existing data on COVID-19 clinical, virological and immunological kinetics, we propose to also discuss earlier immunomodulation to prevent the rise of an auto-inflammatory loop. Such an approach could counter-act the serial immune events leading to hyperinflammation, while immunosuppression should be preferred at a later stage. In order to repurpose immune interventions in COVID-19 patients before oxygen requirement, clinical and biological milestones of the disease evolution (such as dyspnea occurrence, oxygen supplementation requirement, WHO score, surrogate markers of inflammation) should be considered as indirect markers of COVID-19 stage and taken into account in treatment decision. Moreover, as only a small proportion of patients will evolve towards severe forms and ARDS, the development of predictive clinical and biological markers will be crucial in determining which patients should be treated at the early stage of the disease. While the therapeutic arsenal against severe COVID-19 increases, further studies will be needed to refine treatment strategies and characterize the best treatment recipients especially when comparing two drugs beneficial at the same disease stage (i.e., JAK inhibitors and IL-6 antagonists). Lastly, additional studies will be needed to better understand both COVID-19 pulmonary sequelae and the long COVID physiopathology, with the perspective of specific therapeutic approaches. Overall, we believe that a timely approach is crucial to understand COVID-19 pathogenesis and to define therapeutic intervention thresholds, and may be extrapolated to other severe respiratory viral infections, such as influenza infection.
References
Lauer, S. A. et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann. Intern Med. 172, 577–582 (2020).
He, X. et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 26, 672–675 (2020).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Cheng, H.-Y. et al. Contact tracing assessment of COVID-19 transmission dynamics in taiwan and risk at different exposure periods before and after symptom onset. JAMA Intern. Med. 180, 1156–1163 (2020).
Larsen, J. R., Martin, M. R., Martin, J. D., Kuhn, P. & Hicks, J. B. Modeling the Onset of Symptoms of COVID-19. Front Public Health 8, 473 (2020).
Liu, T. et al. The role of interleukin‐6 in monitoring severe case of coronavirus disease 2019. EMBO Mol Med. https://doi.org/10.15252/emmm.202012421 (2020).
Silvin, A. et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell 182, 1401–1418.e18 (2020).
RECOVERY Collaborative Group et al. Dexamethasone in hospitalized patients with Covid-19 - Preliminary Report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2021436 (2020).
Simadibrata, D. M., Calvin, J., Wijaya, A. D. & Ibrahim, N. A. A. Neutrophil-to-lymphocyte ratio on admission to predict the severity and mortality of COVID-19 patients: a meta-analysis. Am. J. Emerg. Med. 42, 60–69 (2021).
Clift, A. K. et al. Living risk prediction algorithm (QCOVID) for risk of hospital admission and mortality from coronavirus 19 in adults: national derivation and validation cohort study. BMJ 371, m3731 (2020).
Lassau, N. et al. Integrating deep learning CT-scan model, biological and clinical variables to predict severity of COVID-19 patients. Nat. Commun. 12, 634 (2021).
Marshall, J. C. et al. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect. Dis. 20, e192–e197 (2020).
Neufeldt, C. J. et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Preprint at https://www.biorxiv.org/content/10.1101/2020.07.21.212639v1 (2020).
Hirano, T. & Murakami, M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity 52, 731–733 (2020).
Winkler, E. S. et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 21, 1327–1335 (2020).
Munster, V. J. et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature 585, 268–272 (2020).
Singh, D. K. et al. SARS-CoV-2 infection leads to acute infection with dynamic cellular and inflammatory flux in the lung that varies across nonhuman primate species. Preprint at https://www.biorxiv.org/content/10.1101/2020.06.05.136481v1 (2020).
Chen, J. et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 84, 1289–1301 (2010).
Iwata-Yoshikawa, N. et al. Acute respiratory infection in human dipeptidyl peptidase 4-transgenic mice infected with middle east respiratory syndrome coronavirus. J. Virol. 93, e01818-18 (2019).
Rockx, B. et al. Comparative pathogenesis of three human and zoonotic SARS-CoV strains in cynomolgus macaques. PLoS One 6, e18558 (2011).
Yao, Z., Zheng, Z., Wu, K. & Junhua, Z. Immune environment modulation in pneumonia patients caused by coronavirus: SARS-CoV, MERS-CoV SARS-CoV-2. Aging 12, 7639–7651 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
Lei, X. et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 11, 3810 (2020).
Combes, A. J. et al. Global absence and targeting of protective immune states in severe COVID-19. Nature 591, 124–130 (2021).
Wathelet, M. G., Orr, M., Frieman, M. B. & Baric, R. S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 81, 11620–11633 (2007).
Matsuyama, T., Kubli, S. P., Yoshinaga, S. K., Pfeffer, K. & Mak, T. W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 27, 3209–3225 (2020).
Carvelli, J. et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature 588, 146–150 (2020).
Galani, I.-E. et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 22, 32–40 (2021).
Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).
Blair, R. V. et al. Acute respiratory distress in aged, SARS-CoV-2–infected african green monkeys but not rhesus macaques. Am. J. Pathol. https://doi.org/10.1016/j.ajpath.2020.10.016 (2020).
Agrati, C. et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ. 27, 3196–3207 (2020).
Zhao, Y. et al. Longitudinal COVID-19 profiling associates IL-1RA and IL-10 with disease severity and RANTES with mild disease. JCI Insight 5, e139834 (2020).
Zhou, Z. et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890.e2 (2020).
Jiang, R.-D. et al. Pathogenesis of SARS-CoV-2 in transgenic mice expressing human angiotensin-converting enzyme 2. Cell 182, 50–58.e8 (2020).
Mathew, D. et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369, eabc8511 (2020).
Woodruff, M. C. et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 21, 1506–1516 (2020).
Kaneko, N. et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell 183, 143–157.e13 (2020).
Suthar, M. S. et al. Rapid generation of neutralizing antibody responses in COVID-19 patients. Cell Rep. Med 1, 100040 (2020).
Aydillo, T. et al. Immunological imprinting of the antibody response in COVID-19 patients. Nat. Commun. 12, 3781 (2021).
Dispinseri, S. et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat. Commun. 12, 2670 (2021).
Lucas, C. et al. Kinetics of antibody responses dictate COVID-19 outcome. medRxiv https://doi.org/10.1101/2020.12.18.20248331 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Zhu, L. et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity 53, 685–696.e3 (2020).
Hoang, T. N. et al. Baricitinib treatment resolves lower-airway macrophage inflammation and neutrophil recruitment in SARS-CoV-2-infected rhesus macaques. Cell 184, 460–475.e21 (2021).
Bronte, V. et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Invest. 130, 6409–6416 (2020).
Patra, T. et al. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 16, e1009128 (2020).
Islam, H., Chamberlain, T. C., Mui, A. L. & Little, J. P. Elevated interleukin-10 levels in COVID-19: potentiation of pro-inflammatory responses or impaired anti-inflammatory action? Front. Immunol. 12, 677008 (2021).
Lindner, H. A., Velásquez, S. Y., Thiel, M. & Kirschning, T. Lung protection vs. infection resolution: interleukin 10 suspected of double-dealing in COVID-19. Front. Immunol. 12, 602130 (2021).
Karki, R. et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168.e17 (2021).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Fan, Y.-Y. et al. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch. Virol. 154, 1093–1099 (2009).
Odak, I. et al. Reappearance of effector T cells is associated with recovery from COVID-19. EBioMedicine 57, 102885 (2020).
Peng, Y. et al. Broad and strong memory CD4 + and CD8 + T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 21, 1336–1345 (2020).
Sekine, T. et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 183, 158–168.e14 (2020).
Shin, H.-S. et al. Immune responses to middle east respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin. Infect. Dis. 68, 984–992 (2019).
Sattler, A. et al. SARS–CoV-2–specific T cell responses and correlations with COVID-19 patient predisposition. J. Clin. Invest. 130, 6477–6489 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629 (2020).
Juno, J. A. et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 26, 1428–1434 (2020).
Zhou, R. et al. Acute SARS-CoV-2 infection impairs dendritic cell and T cell responses. Immunity 53, 864–877.e5 (2020).
Arunachalam, P. S. et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369, 1210–1220 (2020).
Zhao, J., Zhao, J., Van Rooijen, N. & Perlman, S. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. 5, e1000636 (2009).
Yoshikawa, T., Hill, T., Li, K., Peters, C. J. & Tseng, C.-T. K. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 83, 3039–3048 (2009).
Park, S.-J. et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 173, 3844–3854 (2004).
McGregor, R. et al. An autocrine Vitamin D-driven Th1 shutdown program can be exploited for COVID-19. bioRxiv https://doi.org/10.1101/2020.07.18.210161 (2020).
Weiskopf, D. et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 5, eabd2071 (2020).
Chen, Z. & John Wherry, E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 20, 529–536 (2020).
Woolsey, C. et al. Establishment of an African green monkey model for COVID-19 and protection against re-infection. Nat. Immunol. 21, 86–98 (2021).
Peiris, J. S. M. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
De Biasi, S. et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 11, 3434 (2020).
Holter, J. C. et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl Acad. Sci. USA 117, 25018–25025 (2020).
Chakraborty, S. et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat. Immunol. 22, 67–73 (2021).
Larsen, M. D. et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 371, eabc8378 (2021).
Bonaventura, A. et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 21, 319–329 (2021).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128 (2020).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Iba, T., Warkentin, T. E., Thachil, J., Levi, M. & Levy, J. H. Proposal of the definition for COVID-19-associated coagulopathy. J. Clin. Med. 10, 191 (2021).
Schurink, B. et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1, e290–e299 (2020).
Aesif, S. W. et al. Pulmonary pathology of COVID-19 following 8 weeks to 4 months of severe disease: a report of three cases, including one with bilateral lung transplantation. Am. J. Clin. Pathol. https://doi.org/10.1093/ajcp/aqaa264 (2020).
WHO Solidarity Trial Consortium et al. Repurposed antiviral drugs for Covid-19 - interim WHO solidarity trial results. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2023184 (2020).
Monk, P. D. et al. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 9, 196–206 (2021).
Feld, J. J. et al. Peginterferon lambda for the treatment of outpatients with COVID-19: a phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 9, 498–510 (2021).
O’Brien, M. P. et al. Subcutaneous REGEN-COV antibody combination to prevent Covid-19. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2109682 (2021).
Drayman, N. et al. Drug repurposing screen identifies masitinib as a 3CLpro inhibitor that blocks replication of SARS-CoV-2 in vitro. bioRxiv https://doi.org/10.1101/2020.08.31.274639 (2020).
Mizumoto, K., Kagaya, K., Zarebski, A. & Chowell, G. Estimating the asymptomatic proportion of coronavirus disease 2019 (COVID-19) cases on board the Diamond Princess cruise ship, Yokohama, Japan, 2020. Euro Surveill. 25, 2000180 (2020).
Li, C. et al. Estimating the instantaneous asymptomatic proportion with a simple approach: exemplified with the publicly available COVID-19 surveillance data in Hong Kong. Front Public Health 9, 604455 (2021).
D’Alessio, F. R. & Heller, N. M. COVID-19 and myeloid cells: complex interplay correlates with lung severity. J. Clin. Invest. 130, 6214–6217 (2020).
Nishimoto, N. et al. Treatment of rheumatoid arthritis with humanized anti–interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheumatism 50, 1761–1769 (2004).
Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): preliminary results of a randomised, controlled, open-label, platform trial. medRxiv. https://www.medrxiv.org/content/10.1101/2021.02.11.21249258v1.
Shibata, Y. et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15, 557–567 (2001).
Keller, M. J. et al. Effect of systemic glucocorticoids on mortality or mechanical ventilation in patients with COVID-19. J. Hosp. Med 15, 489–493 (2020).
Imai, M. et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl Acad. Sci. USA 117, 16587–16595 (2020).
Liu, S. T. H. et al. Convalescent plasma treatment of severe COVID-19: a propensity score–matched control study. Nat. Med. 26, 1708–1713 (2020).
Libster, R. et al. Early high-titer plasma therapy to prevent severe Covid-19 in older adults. N. Engl. J. Med. 384, 610–618 (2021).
Simonovich, V. A. et al. A randomized trial of convalescent plasma in Covid-19 severe pneumonia. N. Engl. J. Med. 384, 619–629 (2020).
Hueso, T. et al. Convalescent plasma therapy for B-cell-depleted patients with protracted COVID-19. Blood 136, 2290–2295 (2020).
New phase III data shows investigational antibody cocktail casirivimab and imdevimab reduced hospitalisation or death by 70% in non-hospitalised patients with COVID-19. https://www.roche.com/media/releases/med-cor-2021-03-23.htm (2021).
Dougan, M. et al. Bamlanivimab plus etesevimab in mild or moderate Covid-19. N. Engl. J. Med. 385, 1382–1392 (2021).
Planas, D. et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 596, 276–280 (2021).
Robinson PC. et al. Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. The Lancet. Rheumatol. 2, e653–e655 (2020).
Feldmann, M. et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395, 1407–1409 (2020).
Hospices Civils de Lyon Study of the Efficiency and Security of NIVOLUMAB Therapy, Used in Immuno-stimulation, in Hospitalized Obese Individuals at Risk to Evolve Towards Severe Forms of COVID-19 Infection. Multicentric, Paralleled, Randomized, Controlled Trial. https://clinicaltrials.gov/ct2/show/NCT04413838 (2020).
Guan, W.-J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2002032 (2020).
Writing Committee et al. Corticosteroids in patients hospitalised for COVID-19 pneumonia who require oxygen: observational comparative study using routine care data. Clin. Microbiol. Infect. https://doi.org/10.1016/j.cmi.2020.11.035 (2020).
Therapeutic Management. COVID-19 Treatment Guidelines. https://www.covid19treatmentguidelines.nih.gov/therapeutic-management/ (2021).
Salama, C. et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 384, 20–30 (2020).
Hermine, O. et al. Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and Moderate or severe pneumonia: a randomized clinical trial. JAMA Intern. Med. https://doi.org/10.1001/jamainternmed.2020.6820 (2021).
Veiga, V. C. et al. Effect of tocilizumab on clinical outcomes at 15 days in patients with severe or critical coronavirus disease 2019: randomised controlled trial. BMJ 372, n84 (2021).
Rosas, I. O. et al. Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N. Engl. J. Med. 384, 1503–1516 (2021).
Stone, J. H. et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. N. Engl. J. Med. 383, 2333–2344 (2020).
Soin, A. S. et al. Tocilizumab plus standard care versus standard care in patients in India with moderate to severe COVID-19-associated cytokine release syndrome (COVINTOC): an open-label, multicentre, randomised, controlled, phase 3 trial. Lancet Respir. Med. 9, 511–521 (2021).
Salvarani, C. et al. Effect of tocilizumab vs standard care on clinical worsening in patients hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA Intern. Med. https://doi.org/10.1001/jamainternmed.2020.6615 (2020).
Lescure, F.-X. et al. Sarilumab in patients admitted to hospital with severe or critical COVID-19: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 9, 522–532 (2021).
The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group et al. Association between administration of IL-6 antagonists and mortality among patients hospitalized for COVID-19: a meta-analysis. JAMA. https://doi.org/10.1001/jama.2021.11330 (2021).
Therapeutics and COVID-19: living guideline. https://www.who.int/publications-detail-redirect/WHO-2019-nCoV-therapeutics-2021.2 (2021).
Cauchois, R. et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. PNAS 117, 18951–18953 (2020).
Cavalli, G. et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2, e325–e331 (2020).
University Hospital, tours efficacy and safety of ANAkinra during adult “COVID-19“ with aggravating respiratory symptoms: a multicenter open-label controlled randomized trial. https://clinicaltrials.gov/ct2/show/NCT04364009 (2020).
Mariette, X. et al. Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): a randomised controlled trial. Lancet Respir Med. 9, P295-304 (2021).
Caricchio, R. et al. Effect of canakinumab vs placebo on survival without invasive mechanical ventilation in patients hospitalized with severe COVID-19: a randomized clinical trial. JAMA 326, 230–239 (2021).
Somagutta, M. K. R. et al. The safety and efficacy of anakinra, an interleukin-1 antagonist in severe cases of COVID-19: a systematic review and meta-analysis. Infect. Chemother. 53, 221–237 (2021).
Kyriazopoulou, E. et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat. Med. https://doi.org/10.1038/s41591-021-01499-z (2021).
Yan, B. et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci. Immunol. 6, eabg0833 (2021).
Kalil, A. C. et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2031994 (2020).
Guimarães, P. O. et al. Tofacitinib in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 385, 406–415 (2021).
Ltd, KP Kiniksa announces positive results for mavrilimumab phase 2 trial in non-mechanically ventilated severe COVID-19 patients. GlobeNewswire News Room. https://www.globenewswire.com/news-release/2021/04/12/2208144/0/en/Kiniksa-Announces-Positive-Results-for-Mavrilimumab-Phase-2-Trial-in-Non-Mechanically-Ventilated-Severe-COVID-19-Patients.html (2021).
GSK announces results evaluating its investigational monoclonal antibody, otilimab, for the treatment of hospitalised adult patients with COVID-19|GSK. https://www.gsk.com/en-gb/media/press-releases/gsk-announces-otilimab-data-for-treatment-of-covid-19/ (2021).
Pang, J. et al. Efficacy and tolerability of bevacizumab in patients with severe Covid-19. Nature. Communications 12, 814 (2021).
AB science randomized, phase 2 clinical trial to evaluate the safety and efficacy of masitinib combined with isoquercetin, and best supportive care in hospitalized patients with moderate and severe COVID-19. https://clinicaltrials.gov/ct2/show/NCT04622865 (2020).
Assistance Publique - Hôpitaux de Paris comparison of tocilizumab plus dexamethasone vs. dexamethasone for patients with Covid-19. https://clinicaltrials.gov/ct2/show/results/NCT04476979 (2021).
Angus, Derek C. et al. Effect of hydrocortisone on mortality and organ support in patients with severe COVID-19: the REMAP-CAP COVID-19 corticosteroid domain randomized clinical trial. JAMA 324, 317–1329 (2020).
Gupta, S. et al. Association between early treatment with tocilizumab and mortality among critically Ill patients with COVID-19. JAMA Intern Med. https://doi.org/10.1001/jamainternmed.2020.6252 (2020).
WHO recommends life-saving interleukin-6 receptor blockers for COVID-19 and urges producers to join efforts to rapidly increase access. https://www.who.int/news/item/06-07-2021-who-recommends-life-saving-interleukin-6-receptor-blockers-for-covid-19-and-urges-producers-to-join-efforts-to-rapidly-increase-access (2021).
Commissioner, O of the coronavirus (COVID-19) update: FDA authorizes drug for treatment of COVID-19. FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-treatment-covid-19 (2021).
Lo, M. W., Kemper, C. & Woodruff, T. M. COVID-19: complement, coagulation, and collateral damage. J. Immunol. https://doi.org/10.4049/jimmunol.2000644 (2020).
Annane, D. et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. EClinicalMedicine 28, 100590 (2020).
Vlaar, A. P. J. et al. Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19 (PANAMO): an exploratory, open-label, phase 2 randomised controlled trial. Lancet Rheumatol. 2, e764–e773 (2020).
Alexion Provides Update on Phase 3 study of ULTOMIRIS® (ravulizumab-cwvz) in hospitalized patients with severe COVID-19 | Alexion Pharmaceuticals, Inc. https://ir.alexion.com/news-releases/news-release-details/alexion-provides-update-phase-3-study-ultomirisr-ravulizumab (2021).
George, P. M., Wells, A. U. & Jenkins, R. G. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir. Med. 8, 807–815 (2020).
Chaudhary, S., Natt, B., Bime, C., Knox, K. S. & Glassberg, M. K. Antifibrotics in COVID-19 lung disease: let us stay focused. Front. Med. 7, 539 (2020).
Chen, T. et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ 368, m1091 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Matsunaga, N. et al. Clinical epidemiology of hospitalized patients with COVID-19 in Japan: report of the COVID-19 REGISTRY JAPAN. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa1470 (2020).
Fernández-Cruz, A. et al. A retrospective controlled cohort study of the impact of glucocorticoid treatment in SARS-CoV-2 infection mortality. Antimicrob Agents Chemother 64, e01168-20 (2020).
Liu, Z. et al. Low-to-moderate dose corticosteroids treatment in hospitalized adults with COVID-19. Clin. Microbiol Infect. 27, 112–117 (2021).
Rojas-Marte, G. et al. Outcomes in patients with severe COVID-19 disease treated with tocilizumab: a case–controlled study. QJM https://doi.org/10.1093/qjmed/hcaa206 (2020).
Rodríguez-Baño, J. et al. Treatment with tocilizumab or corticosteroids for COVID-19 patients with hyperinflammatory state: a multicentre cohort study (SAM-COVID-19). Clin. Microbiol. Infect. https://doi.org/10.1016/j.cmi.2020.08.010 (2020).
Biran, N. et al. Tocilizumab among patients with COVID-19 in the intensive care unit: a multicentre observational study. Lancet Rheumatol. https://doi.org/10.1016/S2665-9913(20)30277-0 (2020).
Martinez-Sanz, J. et al. Effects of tocilizumab on mortality in hospitalized patients with COVID-19: a multicenter cohort study. Clin. Microbiol. Infect. 27, 238–243 (2021). .
Hung, I. F.-N. et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet 395, 1695–1704 (2020).
Guimarães, P. O. et al. Tofacitinib in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 385, 406–415 (2021).
Weinreich, D. M. et al. Trial investigators. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N. Engl. J. Med. 384, 238–251 (2021).
Author information
Authors and Affiliations
Contributions
L. Plaçais and Q. Richier: study design, data collection, data analysis, data interpretation, writing, figures drawing. O. Hermine, K. Lacombe, X. Mariette, N. Noël: study design, data analysis, writing supervision.
Corresponding authors
Ethics declarations
Competing interests
L.P. and Q.R. did not receive any grants or fees. N.N. reports personal fees from BIOGEN, BMS and Janssen. K.L. reports grants, personal fees, and non-financial support from, MSD; non-financial support and personal fees from Gilead, Janssen and Abbvie; and personal fees from ViiV Healthcare. O.H. reports grants from Celgene, Alexion, AB Science, and Inatherys. X.M. reports grant from Ose and personal fees from BMS, GSK, Janssen, Pfizer, Sanofi and UCB.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Plaçais, L., Richier, Q., Noël, N. et al. Immune interventions in COVID-19: a matter of time?. Mucosal Immunol 15, 198–210 (2022). https://doi.org/10.1038/s41385-021-00464-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-021-00464-w
This article is cited by
-
Remdesivir for the Treatment of COVID-19: A Narrative Review
Infectious Diseases and Therapy (2024)
-
Intranasal influenza-vectored COVID-19 vaccine restrains the SARS-CoV-2 inflammatory response in hamsters
Nature Communications (2023)
