
Abstract
Regulatory T (Treg) cells may participate in mediating a suppressive microenvironment that blunts successful anti-tumor immunotherapy. Recent studies show that CD8+ Treg cells might impede effective immune responses to established tumors. However, there is limited research regarding CD8+ Treg cells in ovarian cancer (OC) patients. Here, we investigated CD8+ Treg cells in OC patients and their in vitro induction. The immunohistochemistry of tumor-infiltrating lymphocytes revealed a significant correlation between the intratumoral CD8+ T cells and the forkhead box p3 (Foxp3)+ cells in the intraepithelial and stromal areas of advanced OC tissues. We examined the expression of Treg markers in CD8+ T cells from the peripheral blood and fresh tumor tissues of OC patients using flow cytometry. Our results indicated an increase in the CD8+ Treg cell subsets of OC patients compared with those in patients with benign ovarian tumors and healthy controls, including an increased expression of CD25, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and Foxp3 and decreased CD28 expression. To demonstrate whether the tumor microenvironment could convert CD8+ effector T cells into suppressor cells, we used an in vitro transwell culturing system. Compared with the CD8+ T cells cultured alone, the CD8+ Treg cells induced in vitro by coculture with SK-OV-3/A2780 showed increased CTLA-4 and Foxp3 expression and decreased CD28 expression. In addition, the in vitro-induced CD8+ Treg cells inhibited naı¨ve CD4+ T-cell proliferation, which was partially mediated through TGF-β1 and IFN-γ. Our study suggests that CD8+ Treg cells were increased in OC patients and could be induced in vitro, which may be the way that tumors limit antitumor immunity and evade immune surveillance.
Similar content being viewed by others


Introduction
Ovarian cancer (OC) is the leading cause of death among gynecological malignancies, which has a complex immune suppressive network that presents major barriers to successful anti-tumor immunotherapy. Despite many therapeutic efforts using new chemotherapies, approximately 80% of patients with advanced OC will have tumor progression or recurrence.1 Immunotherapy is a promising treatment for various types of cancer, including OC.2 However, different types of regulatory T (Treg) cells that lead to immunosuppressive microenvironments in OC blunt successful immune eradication.3
Treg cells consist of functionally diverse subsets of immune suppressive T cells that play a crucial role in the modulation of immune responses and the reduction of deleterious immune activation.4 Based on pre-clinical tumor models, it is well known that Treg cells may participate in the progression of cancer, especially with regard to the ability of Treg cells to promote the development and growth of murine primary tumors.5 However, the influence of Treg cells on tumor progression in the clinic is less clear, with the levels of intratumoral Treg cells correlating with better or worse outcomes depending on the tumor type.6,7
CD4+ Treg cells and CD8+ Treg cells are the two main Treg cell subtypes. Compared with CD4+ Treg cells, CD8+ Treg cells are less well studied. Recent studies indicate that some CD8+ T-cell subsets have regulatory functions in cancer,8,9 autoimmune diseases,10,11,12 and infectious diseases.13,14 Several induced or naturally occurring CD8+ Treg cell subsets have been found to express different cell markers, such as CD25+, forkhead box p3 (Foxp3)+, CD122+, CD103+, and CD28−, depending on the immune environment. Among these markers, CD8+CD28− cells are an important Treg subtype because CD8+CD28− cells exert a suppressive effect on diverse types of diseases, such as diabetes and cancer.15,16 Previous studies have shown an expansion of CD8+CD28− cells in patients with malignancies and that increased CD8+CD28− lymphocyte subsets in cancer patients may be associated with advanced disease stages and poor survival.17 Although several subsets of CD8+ Treg cells have been identified, it is difficult to completely distinguish CD8+ Treg cells from conventional CD8+ T cells because of the lack of a reliable marker.
At present, Foxp3 remains the most specific Treg marker, regardless of its relatively low expression in activated T cells.18 Other widely used markers for Treg cells, such as CD25,19 cytotoxic T-lymphocyte-associated protein 4 (CTLA-4),20 and glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR),21 are also expressed on activated effector T cells. Although these Treg markers are not specific, they are essential tools for defining a subset of Treg cells and are associated with Treg cell functionality. Recent studies have demonstrated that the accumulation of Treg cells was involved in the formation of an immunosuppressive microenvironment in OC patients;22,23 however, there is a limited characterization of CD8+ Treg cells in OC patients. Moreover, it remains unclear whether the OC microenvironment could convert CD8+ effector T cells into suppressive CD8+ Treg cells.
In this study, we demonstrate that subsets of CD8+ Treg cells were increased in OC patients, and the human OC cell line SK-OV-3/A2780 could induce subsets of CD8+ T cell expressed Treg-associated markers and inhibited naı¨ve CD4+ T-cell proliferation, which is mediated partially via TGF-β1 and IFN-γ. To improve antitumor immunotherapy, it is important to understand whether CD8+ Treg cells could be induced within the tumor environment.
Materials and methods
Patients and specimens
The study was approved by the Ethical Committee of the First Affiliated Hospital of Nanjing Medical University (Nanjing, China), and informed consent was obtained from all patients. We studied untreated individuals with OC who were staged according to the International Federation of Gynecology and Obstetrics. We excluded cases who received an operation, radiotherapy, or preoperative chemotherapy before collecting informed consent. Paraffin-embedded specimens and peripheral blood samples were obtained from OC patients and benign ovarian tumor (BOT) patients treated at the First Affiliated Hospital of Nanjing Medical University from 2010 to 2013. Fresh tumor tissues were collected from OC patients and BOT patients treated at the General Hospital of the Nanjing Military Region. Healthy volunteers who underwent a physical examination served as controls. The clinical data of the patients were also collected for analysis. The patient characteristics are displayed in Table 1.
Mononuclear cell isolation
Peripheral blood mononuclear cells (PBMCs) were obtained from blood samples using a Ficoll-Hypaque (TBD, Tianjin, China) density gradient. The tumor tissues were minced into small pieces and then digested using type-IV collagenase, hyaluronidase, and DNase I (all from Sigma, St louis, MO, USA) in serum-free RPMI 1640 medium (Gibco, Gaithersburg, MD, USA) at 37 °C for 1 h under slow rotation. Single-cell suspensions were obtained by filtering the digested tissues through 100 μm cell strainers (BD Falcon, San Jose, CA, USA), and lymphocytes were isolated using Ficoll-Hypaque density gradient centrifugation.
Cell culture
The human OC cell line (SK-OV-3) was purchased from the ATCC (American Type Culture Collection, Manassas, VA, USA) and grown in McCoy’s 5A medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS). Another cell line (A2780) was purchased from KeyGEN (Nanjing, China) and maintained in RPMI 1640 medium supplemented with 10% FBS. The two cell lines were cultured in 5% CO2 at 37 °C.
Immunohistochemistry
The cell populations of CD4+, CD8+ T cells, and Foxp3+ cells in the OC and BOT tissues were determined using immunohistochemistry. Before the immunostaining, the sections were placed in Tris EDTA buffer (10 mM Tris, 1 mM EDTA) at pH 9.0 and heated for 2 min in a pressure cooker. Then, the sections were stained with a panel of the first specific polyclonal antibodies against human CD4 (clone PAB11904, Abnova, 1:200), CD8 (clone PAB11235, Abnova, 1:300), and Foxp3 (clone PAB12685, Abnova, 1:100). The slides were then incubated with secondary antibody labeled polymer horseradish peroxidase for 15 min (Fuzhou Maixin Biotech, Fujian, China). The color development was performed using diaminobenzidine, and the slides were counterstained with hematoxylin, dehydrated and mounted. The expression of the CD4+, CD8+ T cells, and Foxp3+ cells was evaluated using an optical microscope (BX51; Olympus, Tokyo, Japan). The quantitation of the positive cells in the tumor sections was performed using methods as described by Chunling Ma et al.24 Briefly, the number of positive cells was counted in 10 cancer nest areas, 10 cancer stromal areas, and 10 areas that included both cancer nest and stromal areas at a high-power field (400×), and the average number of positive cells per reported field was calculated.
Transwell experiments
CD8+ T cells were isolated from the peripheral blood of healthy adults using a CD8-positive isolation kit (Dynal, Oslo, Norway), and the purities of the isolated cells were >95%. The transwell coculture experiments were conducted in 24-well plates with inner wells (0.4 μm pore size, Corning Costar, Corning, NY, USA) to separate the CD8+ T cells and SK-OV-3/A2780. Freshly isolated CD8+ T cells (2 × 105) were placed in the inner wells containing RPMI 1640 medium with 10% FBS (Gibco). The outer wells contained SK-OV-3/A2780 (4×104) in the same medium or medium alone (for controls). At day 5, the culture supernatant was collected, and the CD8+ T cells were harvested and washed; 1 × 106 cells were collected for the Foxp3, CD25, CD28, CTLA-4, and GITR mRNA analysis, and 2 × 106 cells were collected to detect the expression of Foxp3, CD25, CD28, CTLA-4, and GITR in the CD8+ T cells by flow cytometry, while the rest of the cells were transferred to 96-well plates and stimulated with soluble anti-CD3 (1 mg mL−1; eBioscience, San Diego, CA, USA) plus anti-CD28 (1 mg mL−1; eBioscience) for 6 or 24 h. After the stimulation, the CD8+ T cells were collected and analyzed using real-time PCR, and the supernatants were collected for cytokine detection by ELISA and cytometric bead array (CBA).
Flow cytometry
The fluorochrome-coupled monoclonal antibodies (mAbs) (conjugated with FITC, allophycocyanin (APC), phycoerthrin (PE)) used in this study were: anti-CD8 (FITC), anti-CTLA-4 (APC), anti-CD25 (APC), anti-CD28 (APC), anti-Foxp3 (PE) (all from BD Bioscience, San Jose, CA, USA), and anti-GITR (PE) (eBioscience). Freshly isolated or in vitro cultured cells were washed and incubated with mAbs specific for surface markers, including anti-CD8, anti-CD25, anti-CD28, and anti-GITR in 100 μL PBS for 20 min at room temperature in the dark. The intracellular detection of Foxp3 with anti-Foxp3 and CTLA-4 with anti-CTLA-4 was performed using fixed and permeabilized cells, in accordance with the manufacturer’s instructions. Dead cells were excluded by the forward and side scatter characteristics. The fluorescence labeling was measured with a Gallios Flow cytometer (Beckman Coulter, Fullerton, CA, USA), and the data were analyzed using Kaluza software (Beckman Coulter).
ELISA and cytometric bead array
Transforming growth factor β1 (TGF-β1) in the culture supernatants was tested by ELISA (eBioscience). Interleukin (IL)-10, IL-2, tumor necrosis factor α (TNF-α), and interferon γ (IFN-γ) were measured by flow cytometry with the Human Th1/Th2 Cytokine CBA kit (BD Bioscience).
RNA isolation and real-time PCR
The total RNA of CD8+ T cells from different groups was isolated using the miRNeasy Mini kit (Qiagen, Valencia, CA, USA), following the manufacturer’s instructions and then converted to cDNA. The mRNA expression was determined in an ABI 7500 real-time PCR system (Applied Biosystems/Life Technologies, Foster City, CA, USA) with the use of SYBR Green. The mRNA levels in the CD8+ T cells in each sample were normalized with the relative quantity of β-actin. Each analysis was repeated at least three times. The primers used in this study are shown in Table 2.
Proliferation assays
Naı¨ve CD4+ T cells were isolated from a healthy person with the use of a CD4-positive isolation kit (Dynal, Oslo, Norway) and the Dynabeads FlowComp Human CD45RA kit (Dynal, Oslo, Norway). The CD8+ T cells that were cultured alone or with SK-OV-3/A2780 were added at ratios of 1:0, 1:1, 1:5, 1:10, and 0:1 to naı¨ve CD4+ T cells (1 × 105 cells/well) in 96-well plates. All of the wells were cultured in a final volume of 200 μL with the presence of irradiated PBMCs (5 × 104 cells/well) and 500 ng mL−1 anti-CD3 mAb for 56 h. [3H]-thymidine was added for another 16 h for the determination of proliferation by scintillation counting. The transwell experiments were conducted in 24-well plates as described above. Naı¨ve CD4+ T cells (1 × 105) and 1 × 105 irradiated PBMCs were seeded in the outer wells of a 24-well plate, whereas equal numbers of CD8+ T cells that were cultured with or without SK-OV-3/A2780 were placed in the inner wells. The cells were cultured in medium containing anti-CD3 (500 ng mL−1). The cells in the inner and outer wells were collected separately and transferred to 96-well plates after 56 h of culture. The proliferative response was tested by the addition of [3H] thymidine for another 16 h. For the neutralization experiment, anti-TGF-β1 (1 μg mL−1; R&D Systems, Minneapolis, MN, USA), anti-IFN-γ (2 μg mL−1; BD Bioscience), anti-TNF-α (2 μg mL−1; BD Bioscience), and anti-IL-2 (1 μg mL−1; eBioscience) mAbs were added to the transwell system described above.
Statistical analysis
The results are expressed as mean ± SD. The statistical analysis was performed using the SPSS (Statistical Package for the Social Sciences) 20.0 statistics software (SPSS Inc., Chicago, IL, USA). Differences between the groups and correlations were assessed using the non-parametric Mann–Whitney U-test and the Spearman test, respectively. A P value < 0.05 was considered to be statistically significant.
Results
Prevalence of Foxp3+ cells in the ovarian cancer tissues
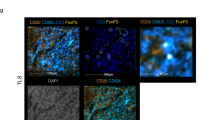
To investigate whether Treg cells were localized at the tumor site, we performed immunohistochemistry to detect the CD4+, CD8+ T cells, and Foxp3+ cells in sections of 41 OC tissues and 12 BOT tissues (Figure 1a). The numbers of positive cells (CD4+, CD8+ T cells, and Foxp3+ cells) per analyzed field were significantly higher in the tissues of the OC patients compared with those in the BOT patients (Figure 1b, P < 0.05). To investigate the clinical significance of increased CD4+, CD8+ T cells, and Foxp3+ cells in the human OC, the tumor stage of the OC patients were analyzed in comparison with the numbers of CD4+, CD8+ T cells, and Foxp3+ cells in the OC tissues. As shown in Figure 1c, the CD8+ T cell and Foxp3+ cell numbers were higher in the OC patients at stage III/IV than those at stage I/II (P < 0.05), while we did not detect any difference in the CD4+ T-cell numbers between the OC patients at stage I/II and III/IV (data not shown). Importantly, among these three subsets of tumor-infiltrating lymphocytes (TILs), the numbers of CD8+ T cells and Foxp3+ cells showed the most significant increase in the advanced OC patients. Thus, we further assessed a possible correlation between the CD8+ T cells and Foxp3+ cells in the intraepithelial and stromal areas of the OC patients at stage III/IV. As a result, we found a significant correlation between the CD8+ T cells and Foxp3+ cells in the intraepithelial and stromal areas of the advanced OC tissues (P < 0.05, r = 0.516 and P < 0.05, r = 0.430, respectively) (Figure 1d). These results suggested that the numbers of both CD8+ T and Foxp3+ cells were highly increased in the advanced OC patients.

The prevalence of Foxp3+ cells in ovarian cancer tissues. (a) Representative images of immunohistochemical staining for CD4, CD8, Foxp3 in consecutive sections of OC tissues and BOT tissues, which were used to analyze the cell numbers. Few CD4+, CD8+ T cells, and Foxp3+ cells were observed in the BOT tissues, while high numbers of CD4+, CD8+ T cells, and Foxp3+ cells were detected in the OC tissues. (b) The numbers of CD4+, CD8+ T cells, and Foxp3+ cells were significantly increased in the OC tissues (n = 41) compared with those in the BOT tissues (n = 12). (c) The numbers of CD8+ T cells and Foxp3+ cells were significantly increased in the OC tissues of patients at stage III/IV (n = 18) compared with those at stage I/II (n = 23). (d) Scatter plots showed a positive correlation of the Foxp3 and CD8 distribution densities in the intraepithelial and stromal compartments of the OC patients, respectively. The average numbers of CD4+, CD8+ T cells, and Foxp3+ cells per field (original magnification: 400×) are shown for each tissue sample. All of the graphs show the mean ± SEM. *P < 0.05, Mann–Whitney U-test and Spearman test.
Expression of Treg markers in the CD8+ T cells from the ovarian cancer patients
The results of the immunohistochemistry demonstrated that the numbers of CD8+ T cells were highly increased in the advanced OC patients. Next, we characterized freshly isolated CD8+ T cells in PBMCs and tissue lymphocytes of the OC patients as well as age-matched BOT patients and healthy individuals as controls (Table 1). We analyzed the expression of Treg markers, including Foxp3, CD25, CD28, CTLA-4, and GITR in the CD8+ T cells. Compared with the BOT tissues, there were increases in the expression of Foxp3 and CTLA-4 and a decrease in the expression of CD28 in the CD8+ T cells from the OC tissues (P < 0.05, P < 0.01, and P < 0.05, respectively). There were no significant differences in the expression of CD25 or GITR in the CD8+ T cells between the OC tissues and BOT tissues (Figure 2). Next, we investigated the expression of Treg markers in the CD8+ T cells from peripheral blood. The proportions of CD25+, Foxp3+, CTLA-4+, and CD28− cells in the CD8+ T cells were increased significantly in the PBMCs from the OC patients compared with those in the BOT patients and healthy controls (P < 0.05, P < 0.01, P < 0.01, and P < 0.01, respectively). However, there was no significant difference in the proportion of GITR+ cells in the CD8+ T cells among the OC patients, BOT patients and healthy controls (Figure 3a and b). Moreover, the level of Foxp3 expression in the CD8+ T cells of the OC patients at stage III/IV was higher than those at stage I/II (Figure 3c). These findings demonstrated that CD8+ Treg cells undergo phenotypic alterations that potentially contribute to their function in OC patients.

The expression of phenotypic Treg markers in the CD8+ T cells from ovarian cancer tissues. (a) The lymphocytes from the OC tissues or the BOT tissues were stained with PE-, APC-, or FITC-labeled mAbs against human CD8, CD25, GITR, Foxp3, CTLA-4, or CD28 molecules, according to the staining protocol described in “Materials and Methods” section. The cumulative data showed an increased expression of Foxp3 and CTLA-4 and a decreased CD28 expression in the CD8+ T cells from the OC patients (n = 14) compared with that in the CD8+ T cells from the BOT patients (n = 12). (b) The gating strategy: the lymphocytes (R1) were gated according to their side- and forward-scatter properties, and the CD8+ T cells (R2) were gated from R1. (c) Representative dot plots of CD8 and Foxp3, CD28, and CTLA-4 staining of the lymphocytes from the OC tissues and BOT tissues. All of the graphs show the mean ± SEM. *P < 0.05, **P < 0.01, Mann–Whitney U-test.

The expression of Treg markers in the CD8+ T cells from the PBMCs of ovarian cancer patients. (a) The PBMCs from the OC patients, BOT patients, and healthy controls were stained with PE-, APC-, or FITC-labeled mAbs against human CD8, CD25, GITR, Foxp3, CTLA-4, or CD28 molecules, according to the staining protocol described in “Materials and Methods” section. The cumulative data showed an increased expression of CD25, Foxp3, and CTLA-4, and a decreased expression of CD28 in the CD8+ T cells from the PBMCs of OC patients (n = 31) compared with that in the CD8+ T cells from the PBMCs of the BOT patients (n = 26) and healthy controls (n = 20). (b) The gating strategy: the lymphocytes (R1) were gated according to their side- and forward-scatter properties, and the CD8+ T cells (R2) were gated from R1. (c) Representative dot plots of CD8 and Foxp3, CD28, CD25, and CTLA-4 staining of the PBMCs from the OC patients, BOT patients, and healthy controls. (d) The cumulative data showed that the percentage of Foxp3+ T cells among the CD8+ T cells from the PBMCs of the OC patients at stage III/IV (n = 24) was higher than that in the patients at stage I/II (n = 7). All of the graphs show the mean ± SEM. *P < 0.05, **P < 0.01, Mann–Whitney U-test.
SK-OV-3 induced phenotypic differentiation of CD8+ T cells in a transwell coculture system
To test whether CD8+ Treg cells can be induced in vitro by SK-OV-3, we first evaluated the phenotypes of the CD8+ T cells cultured with or without SK-OV-3 using a transwell coculture system. As shown in Figure 4, compared with the CD8+ T cells cultured without SK-OV-3, the expression of Foxp3 and CTLA-4 was increased and the CD28 expression was decreased in the CD8+ T cells cultured with SK-OV-3 (both P < 0.001). There was no significant difference in the expression of CD25 or GITR between the CD8+ cells cultured with or without SK-OV-3. Our results showed that the frequency of Foxp3+, CTLA-4+, and CD28− cells gated on the CD8+ T cells was increased in the transwell culture system with SK-OV-3.

The SK-OV-3 cells induce a phenotypic differentiation of the CD8+ T cells in a transwell coculture system. (a) The expression of CD25, GITR, CTLA-4, Foxp3, and CD28 mRNA in the CD8+ T cells cultured with or without SK-OV-3 was analyzed by quantitative real-time PCR. (b) The CD8+ T cells in the transwell coculture system with SK-OV-3 expressed higher levels of the CTLA-4 and Foxp3 molecules and lower levels of CD28 than that in the CD8+ T cells cultured alone. The expression of CD25 and GITR was low, and there was no significant difference between the CD8+ T cells cultured with and without SK-OV-3. (c) Representative dot plots of the CD8 and Foxp3, CD28, and CTLA-4 staining of the CD8+ T cells cultured with or without SK-OV-3. *P < 0.05, **P < 0.01.
Cytokine expression profile of in vitro-differentiated CD8+ Treg cells
In addition to the phenotypic changes of the CD8+ T cells in the transwell coculture system with SK-OV-3/A2780, the CD8+ T cells also showed a vigorous TGF-β1, IFN-γ, TNF-α, and IL-2 response with the stimulation of anti-CD3/CD28 mAbs for 6 or 24 h, while the CD8+ T cells cultured without SK-OV-3/A2780 showed poor TGF-β1, IFN-γ, TNF-α, and IL-2 production (Figure 5a). The results of the real-time PCR also indicated that the CD8+ T cells cultured with SK-OV-3 expressed higher TGF-β1, IFN-γ, TNF-α, and IL-2 mRNA after the anti-CD3/CD28 mAbs stimulation for 6 or 24 h compared with that in the CD8+ T cells cultured without SK-OV-3 (Figure 5b). IL-10 is important for the suppressive activity of Treg cells. Although the real-time PCR analysis indicated that the IL-10 mRNA expression in the CD8+ T cells cultured with SK-OV-3 was higher than that in the CD8+ T cells cultured without SK-OV-3; we found no or little IL-10 secretion from the CD8+ T cells cultured with or without SK-OV-3 after stimulation with the anti-CD3/CD28 mAbs (data not shown). The analyses of the phenotypes and cytokine secretion indicated that CD8+ Treg cells could be induced from peripheral blood CD8+ T cells by coculture with SK-OV-3.

The cytokine expression profile of the in vitro-differentiated CD8+ Treg cells. (a) The production of TGF-β1, IL-10, IFN-γ, IL-2, and TNF-α in the CD8+ T cells cultured with or without SK-OV-3. The supernatants were collected after the CD8+ T cells were stimulated with anti-CD3/CD28 stimulation for 6 or 24 h. TGF-β1 was detected by ELISA, and IL-10, IFN-γ, IL-2, and TNF-α were detected by CBA. (b) The real-time RT-PCR analysis of the TGF-β1, IL-10, IFN-γ, IL-2, and TNF-α mRNA expression in the CD8+ T cells cultured with or without SK-OV-3. The total RNA was isolated from the CD8+ T cells stimulated with anti-CD3/CD28 mAbs for 6 or 24 h. **P < 0.01.
In vitro-differentiated CD8+ Treg cells suppress the proliferation of naı¨ve CD4+ T cells
In addition to examining the phenotypes and cytokine expression profiles of the CD8+ T cells cultured with SK-OV-3, we explored the function of these CD8+ T cells. As shown in Figure 6a, we found that the CD8+ T cells cultured with SK-OV-3 suppressed the naı¨ve CD4+ T-cell proliferation in the presence of an anti-CD3 mAb in a dose-dependent manner, while the CD8+ T cells cultured without SK-OV-3 did not suppress the naı¨ve CD4+ T-cell proliferation. Our results indicated that CD8+ T cells cultured with SK-OV-3 exerted a potent immunosuppressive effect on the naı¨ve CD4+ T-cell proliferation. To determine whether cell-to-cell contact or soluble factors were required for the suppressive activity of these CD8+ T cells, we performed direct coculture and transwell experiments. The CD8+ T cells cultured with SK-OV-3 inhibited the naı¨ve CD4+ T-cell proliferation, not only in the direct coculture but also in the transwell system (Figure 6b). With the use of neutralizing anti-TGF-β1 and IFN-γ mAbs, we found that the suppressive effect of the CD8+ T cells cultured with SK-OV-3 could be partially blocked, while the blockade of TNF-α and IL-2 did not affect the suppressive ability of the CD8+ T cells cultured with SK-OV-3 (Figure 6c). Our results showed that the suppressive ability of the CD8+ T cells cultured with SK-OV-3 was mediated at least in part by soluble factors, and the immunosuppression could be alleviated by anti-TGF-β1 and IFN-γ mAb. The same analysis was performed in the transwell coculture system using another OC cell line, A2780, and CD8+ T cells, which revealed that A2780 and SK-OV-3 could induce CD8+ T cells in an in vitro transwell coculture system, with similar Treg-associated characteristics (Supplementary Figures S1–S3).

In vitro differentiated CD8+ T cells suppressed the proliferation of naïve CD4+ T cells. (a) The CD8+ T cells cultured with SK-OV-3 suppressed naïve CD4+ T-cell proliferation in a dose-dependent manner with the stimulation by anti-CD3 mAb. Conversely, the CD8+ T cells cultured alone did not have the suppressive activity. The proliferative response was determined by [3H] thymidine uptake. (b) Soluble factors were partially required for the CD8+ T cells cultured with SK-OV-3 to exert their suppressive effects. In the transwell system, equal numbers of naïve CD4+ T cells were cultured in the outer wells, and the CD8+ T cells cultured with/without SK-OV-3 were cultured in the inner wells. (c) The suppressive effect of the CD8+ T cells cultured with SK-OV-3 could be partially blocked with the use of neutralizing anti-TGF-β1 and IFN-γ mAbs, while the blockade of TNF-α and IL-2 did not affect the suppressive ability of the CD8+ T cells cultured with SK-OV-3. C.P.M. is an abbreviation for counts per minute. The data are presented as the mean ± SEM of three independent experiments. *P < 0.05.
TGF-β1 may participate in the induction of CD8+ Treg cells in an in vitro transwell coculture system
We detected the expression of cytokines released in the culture medium that may contribute to the induction of CD8+ Treg cells. As shown in Supplementary Figure S4, we found that compared with the CD8+ T cells cultured without the SK-OV-3 system, the CD8+ T cells cultured with the SK-OV-3 system secreted a higher level of TGF-β1 in the medium (P < 0.01), while we found no or little IL-10, IFN-γ, TNF-α, IL-2, IL-4, and IL-6 in the medium (data not shown). The higher level of TGF-β1 in our study system may participate in the induction of CD8+ Treg cells.
Discussion
The presence of TILs in the tumor environment indicates that host immune cells can respond to tumor antigens.25,26 For successful anti-tumor immunotherapy, it is important for us to understand the roles of different subtypes of TILs in the immunosuppressive microenvironment. An immunosuppressive environment still forms at the tumor site regardless of the infiltration of the effector TILs that respond to tumor antigens by exerting their cytotoxic effect and releasing soluble factors.27 Tumor immune tolerance is mainly because of the immunosuppressive effect of Treg cells on anti-tumor immunity.28 Treg cells induce immune tolerance via their suppressive effect on self- and non-self-antigens and contribute to the inhibition of autoimmunity. However, they can also impair anti-tumor immunity.29 Until now, Foxp3 has been the most specific marker to distinguish Treg cells from T cells. Our results suggested that the numbers of CD4+, CD8+ T cells, and Foxp3+ cells were highly increased in the OC tissues compared with those in the BOT tissues (Figure 1). Due to the cytotoxic T-lymphocyte activity of CD8+ T cells, researchers pay more attention to their antitumor functions in immunotherapy.30 It has also been reported that the number of CD8+ T cells is selectively decreased in colorectal cancer patients.31 However, our results showed that the numbers of tumor-infiltrating CD8+ T cells were positively correlated with the tumor stage.
Additionally, we found that a high tumor density of Foxp3+ cells in the OC patients was associated with a poor clinical outcome, indicating that the presence of Treg cells might play a key role in OC development. Importantly, we revealed a significant correlation between the intratumoral CD8+ T cells and the Foxp3+ cells in the intraepithelial and stromal areas of advanced OC tissues. Based on these data, we hypothesized that the growth of tumors in OC patients induces the generation of CD8+ Treg cells. Tumors could secrete soluble factors that promote the induction, expansion, and recruitment of Treg cells to the tumor microenvironment. Therefore, tumors contribute to the generation and expansion of Treg cells in the tumor microenvironment.18 The phenotype and function of CD8+ Treg cells are different in diverse tumor models.32
To confirm whether CD8+ T cells in OC patients constitute a subtype of Treg cells, we compared the expression of Treg markers in the blood and tissue CD8+ T cells in the OC patients with that in BOT patients and healthy volunteers. The CD8+ T cells from the PBMCs of OC patients had an increased expression of CD25, Foxp3, and CTLA-4 and a decreased expression of CD28, while the CD8+ T cells in the tumor tissues of the OC patients showed an increased expression of Foxp3 and CTLA-4 and a decreased CD28 expression. Our study demonstrated that the CD8+ T-cell subsets with the phenotypic features of Treg cells were expanded in the tumor tissues and peripheral blood of patients with primary OC. We also found that the expression of Foxp3 in the CD8+ T cells was positively correlated with the tumor stage in the OC patients. These results indicated that CD8+Foxp3+ Treg cells may be involved in the progression of OC, and Foxp3 expression in CD8+ T cells may be regarded as a marker to predict the clinical outcome of OC patients.
There are three modes of Treg cells accumulation within the tumor environment: increased migration, preferential Treg cells expansion, and de novo conversion of Foxp3− T cells into Treg cells.33 To explore whether the tumor microenvironment could convert CD8+ effector T cells into suppressor cells, we established an in vitro transwell culturing system. Our results showed that CD8+ T cells in the transwell coculture system shared similar phenotypic features with CD8+ T cells in OC tissues, namely, increased expression of the Treg cell-associated molecules Foxp3 and CTLA-4 and decreased CD28 expression. The cytokine profile analysis showed that the CD8+ T cells cultured with SK-OV-3 secreted TGF-β1, IFN-γ, TNF-α, and IL-2, but no or little IL-4 and IL-10. TGF-β plays a vital role in the ability of Treg cells to exert their suppressive function.34,35 Recent studies indicate that IFN-γ may be immunosuppressive, and CD8+ Treg cells cannot be induced in IFN-γ R−/− mice.36 TNF-α is known for its ability to desensitize T cells to T-cell receptor activation, and there is increasing evidence that TNF-α contributes to a loss of CD28 expression.37
Importantly, CD8+ T cells in the transwell coculture system had a potent suppressive activity that strongly suppressed the proliferation of the naı¨ve CD4+ T cells (Figure 6a). The possible mechanisms by which CD8+ Treg cells exert their suppressive effect may vary depending on diverse immune reactions. Recent studies indicate that the suppressive activity of Treg cells is mainly mediated through soluble factors, such as IL-10 and/or TGF-β, or cell–cell contact.38,39 In our study, the CD8+ T cells cultured with SK-OV-3 exerted their suppressive effects not only in the direct coculture but also in the transwell system, which could be blocked by anti-TGF-β1 and IFN-γ mAbs (Figure 6c), implying that the suppressive activity of these cells is partially mediated through TGF-β1 and IFN-γ. These results indicated that SK-OV-3-induced subsets of CD8+ T cells expressed Treg markers and acquired the capacity to suppress the proliferative response of naı¨ve CD4+ T cells. We also found that the CD8+ T cells cultured with the SK-OV-3 transwell system secreted a higher level of TGF-β1 in the medium (Supplementary Figure S4), implying that TGF-β1 may contribute to the induction of CD8+ Tregs in an in vitro transwell coculture system. TGF-β signals are important for Tregs, and they could convert CD4+CD25− T cells into CD4+ Tregs through upregulating Foxp3 expression.40 However, the detailed mechanism/signals that are involved in the induction of CD8+ Tregs need further study.
In conclusion, our findings provide new insights into the composition and function of CD8+ T cells in OC patients, which may contribute to the development of T-cell-based adoptive immunotherapy for OC. SK-OV-3 could convert CD8+ T cells into CD8+ Treg cells which may partially promote tumor progression in OC.
Competing Interests
The authors declare no financial or commercial conflict of interest.
References
Luvero D, Milani A, Ledermann JA . Treatment options in recurrent ovarian cancer: latest evidence and clinical potential. Ther Adv Med Oncol 2014; 6: 229–239.
Zsiros E, Tanyi J, Balint K, Kandalaft LE . Immunotherapy for ovarian cancer: recent advances and perspectives. Curr Opin Oncol 2014; 26: 492–500.
Yigit R, Massuger LF, Figdor CG, Torensma R . Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol Oncol 2010; 117: 366–372.
Facciabene A, Motz GT, Coukos G . T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012; 72: 2162–2171.
Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ et al. Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann Surg Oncol 2006; 13: 1252–1258.
Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z et al. Prognostic significance of FOXP3+ tumor infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res 2014; 16: 432.
Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 2009; 27: 186–192.
Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Research 2007; 13: 6947–6958.
Li J, Huang ZF, Xiong G, Mo HY, Qiu F, Mai HQ et al. Distribution, characterization, and induction of CD8+ regulatory T cells and IL-17-producing CD8+ T cells in nasopharyngeal carcinoma. J Transl Med 2011; 9: 189.
Alvarez Arias DA, Kim HJ, Zhou P, Holderried TA, Wang X, Dranoff G et al. Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol Res 2014; 2: 207–216.
Notley CA, McCann FE, Inglis JJ, Williams RO . ANTI-CD3 therapy expands the numbers of CD4+ and CD8+ Treg cells and induces sustained amelioration of collagen-induced arthritis. Arthritis Rheum 2010; 62: 171–178.
Tardito S, Negrini S, Conteduca G, Ferrera F, Parodi A, Battaglia F et al. Indoleamine 2,3 dioxygenase gene polymorphisms correlate with CD8+ Treg impairment in systemic sclerosis. Human Immunol 2013; 74: 166–169.
Holderried TA, Lang PA, Kim HJ, Cantor H . Genetic disruption of CD8+ Treg activity enhances the immune response to viral infection. Proc Natl Acad Sci U S A 2013; 110: 21089–21094.
Zou Q, Wu B, Xue J, Fan X, Feng C, Geng S et al. CD8+ Treg cells suppress CD8+ T cell-responses by IL-10-dependent mechanism during H5N1 influenza virus infection. Eur J Immunol 2014; 44: 103–114.
Yarde DN, Lorenzo-Arteaga K, Corley KP, Cabrera M, Sarvetnick NE . CD28(−) CD8(+) T cells are significantly reduced and correlate with disease duration in juveniles with type 1 diabetes. Human Immunol 2014; 75: 1069–1074.
Chen C, Chen D, Zhang Y, Chen Z, Zhu W, Zhang B et al. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int Immunopharmacol 2014; 18: 255–261.
Song G, Wang X, Jia J, Yuan Y, Wan F, Zhou X et al. Elevated level of peripheral CD8(+)CD28(-) T lymphocytes are an independent predictor of progression-free survival in patients with metastatic breast cancer during the course of chemotherapy. Cancer Immunol Immunother 2013; 62: 1123–1130.
Wang RF . CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Human Immunol 2008; 69: 811–814.
McNeill A, Spittle E, Backstrom BT . Partial depletion of CD69low-expressing natural regulatory T cells with the anti-CD25 monoclonal antibody PC61. Scand J Immunol 2007; 65: 63–69.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008; 322: 271–275.
Uraushihara K, Kanai T, Ko K, Totsuka T, Makita S, Iiyama R et al. Regulation of murine inflammatory bowel disease by CD25+ and CD25- CD4+ glucocorticoid-induced TNF receptor family-related gene+ regulatory T cells. J Immunol 2003; 171: 708–716.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10: 942–949.
Napoletano C, Bellati F, Landi R, Pauselli S, Marchetti C, Visconti V et al. Ovarian cancer cytoreduction induces changes in T cell population subsets reducing immunosuppression. J Cell Mol Med 2010; 14: 2748–2759.
Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Wevers E et al. Tumor-infiltrating gammadelta T lymphocytes predict clinical outcome in human breast cancer. J Immunol 2012; 189: 5029–5036.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003; 348: 203–213.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102: 18538–18543.
Zou W . Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5: 263–274.
Steer HJ, Lake RA, Nowak AK, Robinson BW . Harnessing the immune response to treat cancer. Oncogene 2010; 29: 6301–6313.
Sakaguchi S . Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 2004; 22: 531–562.
Fassnacht M, Lee J, Milazzo C, Boczkowski D, Su Z, Nair S et al. Induction of CD4(+) and CD8(+) T-cell responses to the human stromal antigen, fibroblast activation protein: implication for cancer immunotherapy. Clin Cancer Research 2005; 11: 5566–5571.
Jang TJ . Progressive increase of regulatory T cells and decrease of CD8+ T cells and CD8+ T cells/regulatory T cells ratio during colorectal cancer development. Korean J Pathol 2013; 47: 443–451.
Jarnicki AG, Lysaght J, Todryk S, Mills KH . Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol 2006; 177: 896–904.
Oleinika K, Nibbs RJ, Graham GJ, Fraser AR . Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol 2013; 171: 36–45.
O’Garra A, Vieira P . Regulatory T cells and mechanisms of immune system control. Nat Med 2004; 10: 801–805.
Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA . Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol 2004; 172: 5213–5221.
Chen ML, Yan BS, Kozoriz D, Weiner HL . Novel CD8+ Treg suppress EAE by TGF-beta- and IFN-gamma-dependent mechanisms. Eur J Immunol 2009; 39: 3423–3435.
Bryl E, Vallejo AN, Weyand CM, Goronzy JJ . Down-regulation of CD28 expression by TNF-alpha. J Immunol 2001; 167: 3231–3238.
Field EH, Kulhankova K, Nasr ME . Natural Tregs, CD4+CD25+ inhibitory hybridomas, and their cell contact dependent suppression. Immunol Res 2007; 39: 62–78.
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204: 1257–1265.
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N et al. Conversion of peripheral CD4+CD25-naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198: 1875–1886.
Acknowledgements
This study was supported by the Natural Science Foundation of China (Grant No. 81272324, 81371894) and Key Laboratory for Medicine of Jiangsu Province of China (Grant No. XK201114).
Author information
Authors and Affiliations
Additional information
Supplementary information of this article can be found on Cellular & Molecular Immunology website: http://www.nature.com/cmi.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if thematerial is not included under the Creative Commons license, users will need to obtain permission fromthe license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Zhang, S., Ke, X., Zeng, S. et al. Analysis of CD8+ Treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol 12, 580–591 (2015). https://doi.org/10.1038/cmi.2015.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2015.57
Keywords
This article is cited by
-
Induced pluripotent stem cell-derived natural killer cells gene-modified to express chimeric antigen receptor-targeting solid tumors
International Journal of Hematology (2021)
-
Regulatory lymphocytes: the dice that resolve the tumor endgame
Applied Cancer Research (2020)
-
Distribution and functions of γδ T cells infiltrated in the ovarian cancer microenvironment
Journal of Translational Medicine (2019)
-
Efficacy of PD-1 blockade in cervical cancer is related to a CD8+FoxP3+CD25+ T-cell subset with operational effector functions despite high immune checkpoint levels
Journal for ImmunoTherapy of Cancer (2019)
-
T cell senescence and CAR-T cell exhaustion in hematological malignancies
Journal of Hematology & Oncology (2018)
