
Abstract
Cytomegaloviruses (CMVs), members of the herpesvirus family, have evolved a variety of mechanisms to evade the immune response to survive in infected hosts and to establish latent infection. They effectively hide infected cells from the effector mechanisms of adaptive immunity by eliminating cellular proteins (major histocompatibility Class I and Class II molecules) from the cell surface that display viral antigens to CD8 and CD4 T lymphocytes. CMVs also successfully escape recognition and elimination of infected cells by natural killer (NK) cells, effector cells of innate immunity, either by mimicking NK cell inhibitory ligands or by downregulating NK cell-activating ligands. To accomplish these immunoevasion functions, CMVs encode several proteins that function in the biosynthetic pathway by inhibiting the assembly and trafficking of cellular proteins that participate in immune recognition and thereby, block their appearance at the cell surface. However, elimination of these proteins from the cell surface can also be achieved by perturbation of their endosomal route and subsequent relocation from the cell surface into intracellular compartments. Namely, the physiological route of every cellular protein, including immune recognition molecules, is characterized by specific features that determine its residence time at the cell surface. In this review, we summarize the current understanding of endocytic trafficking of immune recognition molecules and perturbations of the endosomal system during infection with CMVs and other members of the herpesvirus family that contribute to their immune evasion mechanisms.
Similar content being viewed by others


Introduction
To evade host immune surveillance, herpesviruses have evolved a variety of mechanisms that provide them with the capability of lifelong survival in infected hosts in the form of latent infection with the ability to reactivate when the immune surveillance is compromised.1 These mechanisms are based on the synthesis of viral gene products that specifically interrupt the expression, either the synthesis or intracellular processing, of various effector molecules of the innate and adaptive immune response.2 A majority of these gene products target either cellular machineries that present viral antigens to major effector cells with specific immunity (CD8 T cells, CD4 T cells and natural killer (NK) T cells)2 or molecules that contribute to immune recognition by major effector cells that contribute to innate immunity (NK cells).3
Cytomegaloviruses (CMVs), members of the herpesvirus family, encode a relatively large number of gene products that interfere with primary effector mechanisms. They disrupt the presentation of antigen to CD8 and CD4 T lymphocytes and attack ligands that activate NK cells.4 To achieve this goal, they specifically target immune recognition molecules at the earliest stages of assembly and intracellular trafficking in the biosynthetic pathway.2,5 However, interference with later stages of their intracellular life is poorly understood, although it is well known that CMVs perturb many cellular processes, especially the vesicular system. Every cellular protein that resides in the membranous system of the cell has specific features that determine its intracellular localization and lifetime. Thus, perturbation of the endosomal route of cellular proteins, including molecules that participate in immune recognition, may result in distinct cellular routing.
In this review, we will focus on CMV remodeling of the endosomal system and the consequent perturbation of endocytic trafficking of major immune recognition molecules, which can contribute to immune evasion mechanisms. First, we will provide a brief overview of our current understanding of the physiology of endocytic trafficking, then we will describe known CMV effects on endocytic routes of immune recognition molecules and finally, we will give a brief overview of the mechanisms used by other herpesviruses.
The endocytic pathway
Despite intensive research in the last two decades, the endosomal system and endocytic routes remain poorly understood. In fact, as increasing data are obtained for that complex pathway, the number of unresolved issues has also risen. The size and dynamics of the endocytic pathway differ significantly in various cell lines due to an extremely high degree of plasticity. Figure 1 shows a schematic of the endosomal system and endosomal routes relevant to the discussion of immune evasion by endosomal perturbation.

Schematic illustration of the endosomal system and endocytic pathways. Endocytic carriers and endosomal compartments are localized in cellular areas that are organized by the actin cytoskeletal network (light blue area) and microtubular network (darker blue). The dark blue area represents the nucleus. Basic types of endosomal invaginations indicate routes of endosomal uptake: DTI, deep tubular invaginations; CDE, clathrin-dependent endocytosis; CIE, clathrin-independent endocytosis; MP, macropinocytosis. Yellow compartments present various stages of endosomal maturation: EE, early endosomes; EE/SE, early/sorting endosomes; JRC, juxtanuclear recycling compartment; MVB, multivesicular bodies; LE, late endosomes; LY, lysosomes. Endosomal recycling routes are labeled in red: rrp, rapid recycling pathway; frp, fast recycling pathway; srp, slow recycling pathway; lerp, late endosomal recycling pathway.
Internalization and internalization rate
Internalization represents the loss of cell surface molecules as a net result of the combined effects of endocytosis (entrance of molecules from the cell surface into the cell interior) and recycling (return of endocytosed molecules back to the cell surface).6 Thus, the extent and rate of internalization is determined based on both the rates of endocytosis and recycling. The endocytic activity of the plasma membrane is extremely high. The equivalent of the cell surface is internalized one to five times per hour,7 which indicates that endocytic and recycling activity are highly dynamic and well coordinated. Therefore, if a membrane protein has a constitutively higher endocytic rate than the recycling rate, more protein will accumulate in the intracellular pool. A decrease in the recycling rate (i.e., by remodeling of the endocytic route during viral infection) will result in a relocation of plasma membrane protein to the intracellular site where the trafficking rate is the slowest.
Endocytic pathways and endocytic rates
As mentioned previously, endocytosis can be described as a movement of membranes from the cell surface into an internal compartment by the formation of various types of endocytic invaginations and endocytic carriers. The endocytic rate is not uniform for the entire plasma membrane and depends on the membrane composition and molecular machinery used to form the endocytic invaginations and endocytic carriers. The most frequently discussed endocytic pathways involve clathrin-dependent endocytosis, clathrin-independent endocytosis, macropinocytosis and endocytosis through deep tubular invaginations (Figure 1).
A significant fraction of the plasma membrane concentrates membrane proteins that contain specific cytoplasmic motifs (clathrin-dependent endocytic motifs such as the tyrosine-based motif, the di-leucine-based motif, NPXY and mono/multi-ubiquitinylation). These motifs are recognized by adaptor proteins (AP-2), leading to rapid endocytosis by the mobilization of complex machinery containing clathrin to form clathrin-coated endocytic carriers.8 At every moment, 1%–2% of the plasma membrane contains clathrin-coated pits, and approximately 50% of the plasma membrane is internalized each hour through this endocytic route.7 This pathway is used to endocytose many proteins and receptors (i.e., transferrin receptor (TfR), epidermal growth factor receptor (EGFR)).
Membrane proteins that do not have clathrin-dependent endocytic motifs are captured into clathrin-independent endocytic invaginations and carriers that are constitutively formed at the plasma membrane. A portion of these carriers are associated with engagement of the protein caveolin (caveolae-dependent endocytosis). Another portion is associated with the activity of the small GTPase Arf6 (known as the Arf6-associated pathway), whereas some carriers are formed by tubular invaginations that do not associate with Arf6, but require the activity of the small GTPase cdc42, Arf1 and the actin cytoskeleton, known as the cdc42-dependent pathway or GEEC (glycosylphosphatidyl anchored proteins enriched endosomal compartments) pathway.9,10,11,12,13,14 Additionally, the plasma membrane contains poorly characterized deep tubular invaginations15 that ensure the endocytic uptake and cycling of many membrane proteins.16
With increasing data on clathrin-independent endocytosis, it is becoming apparent that there are multiple and overlapping routes for constitutive membrane uptake, and thus, the classification of clathrin-independent endocytic routes is constantly changing. Membrane deformations and the development of endocytic invaginations depend on the lipid composition of the involved membrane region, engagement of effector molecules and association with the actin cytoskeleton. For example, membrane proteins are organized in different types of microdomains (sphingomyelin organized microdomains,17 tetraspanin-rich microdomains18) and nanoclusters of various sizes and composition.19 The composition of membrane microdomains determines the mode of membrane deformation and development of endocytic invaginations, including the engagement of various components of the endocytic machinery that are available in the cytosol. Large lipid-organized areas of the membrane (lipid rafts) will certainly require a distinct machinery for deformation, budding and scission compared with lipid-disordered areas. Therefore, several different endocytic rate constants of constitutive endocytic uptake can be expected based on the heterogeneity of the membrane composition.
In addition to endocytic uptake by clathrin-dependent and clathrin-independent mechanisms, a significant portion of the plasma membrane is also internalized by macropinocytosis (Figure 1), membrane ruffles that are formed at particular types of membrane subdomains.20 These ruffles also mediate uptake of extracellular fluid and cargo molecules in the fluid (i.e., dextran and albumin).
Taken together, it is clear that the overall endocytic rate is a composite of several endocytic activities that occur simultaneously at the plasma membrane: the rate of clathrin-dependent endocytic uptake, rates of constitutive membrane uptake via clathrin-independent endocytic carriers and deep tubular invaginations, and the rate of macropinocytic uptake.
Early endosomal route
After endocytic uptake, clathrin-coated endocytic carriers are rapidly uncoated and fuse together. Their phospholipid composition changes (i.e., acquire phosphatidylinositol 3-phosphate) and they bind the small GTPase Rab5 and early endosomal antigen 1 (EEA1), are tethered to microtubules and become classical early endosomes (Figure 1).21 The same events also characterize the route of clathrin-independent endocytic vesicles. However, it seems that the latter undergo several pre-early endosomal maturation events, including the formation of recycling carriers.9,10,11,12,13 These actin network-associated events are poorly characterized and represent an area of intensive research. After maturation to the point of acquisition of EEA1, non-clathrin-derived endocytic carriers fuse with classical early endosomes and mix cargo molecules with those being shuttled through the clathrin-dependent route.9,10,11,12,13,21,22 Early endosomes attached to microtubules travel towards the cell center while accepting new cargo vesicles and releasing tubular extensions.22 By enlargement, classical early endosomes mix cargo materials and form several membrane domains of maturation.
Along the entire early endocytic route, endosomal membranes form recycling endosomal domains that tubulate and generate recycling endocytic carriers (Figure 1).23 The recycling of membranes starts early after endocytic uptake, just before the stage of classical early endosomes. This poorly characterized pathway of recycling, known as rapid recycling,24 occurs in the cortical area and requires the actin cytoskeleton.16 Upon fusion into classical early endosomes that travel towards the cell center, early endosomal membranes continuously generate recycling domains that return cargo (i.e., TfR and EGFR) back to the cell surface. This route, known as the fast recycling route, returns both clathrin-dependent and clathrin-independent cargo molecules, although it appears that at least some clathrin-independent cargo molecules (i.e., major histocompatibility (MHC) I proteins) are recycled via tubular endosomes that are distinct from classical TfR-loaded recycling carriers.25,26 Finally, early endosomes that reach the cell center segregate a substantial fraction of the membrane cargo into endolysosomal domains that mature into late endosomes;21 the rest is constitutively transported into enlarged tubular endosomes that concentrate around the cell center, known as the juxtanuclear recycling compartment (JRC).21,22 The JRC cargo molecules are slowly recycled back to the plasma membrane (slow recycling pathway, Figure 1).
Recycling from the early endosomal system occurs continuously at a rate that counteracts the high rate of endocytic uptake. If an equivalent of the plasma membrane is endocytosed one to five times per hour, the majority of the endocytosed membranes should be returned via recycling at a similar rate because only ∼5% of the plasma membrane is newly synthesized each hour.7 Thus, the proper functioning and integrity of the recycling system is a key regulator of the composition of membrane organelles and the cellular distribution of membrane proteins.
Late endosomal route
During their movement along microtubules, early endosomes enlarge by absorbing incoming early endosomal carriers.21,27 Simultaneously to the constant generation of recycling domains, early endosomes generate an endolysosomal domain that is characterized by inward budding and the formation of intraluminal vesicles (Figure 1). This domain contributes to the formation of vacuolar early endosomes, often termed sorting endosomes, which mature into multivesicular bodies (MVBs) and either exchange cargo with late endosomes or fuse with late endosomal membranes in a process that remains poorly characterized.21 Late endosomes are considered a highly dynamic network of vesicular endosomal membranes that mix cargo molecules and deliver them either to lysosomes for degradation or to the trans-Golgi network (TGN) for utilization in the secretory pathway.21,28 A fraction of multivesicular endosomes either develop into specialized lysosome-related organelles29,30 or constitutively reach the cell surface and recycle late endosomal cargo to the plasma membrane31 (lerp, late-endosomal recycling pathway, Figure 1). Intraluminal vesicles are released in the extracellular milieu as exosomes during the process known as exocytosis.21,32
Regulation of endosomal dynamics
Endosomal carriers are formed by membrane deformations caused by changes in the membrane lipid composition (catalyzed by lipid-modifying enzymes) with the assistance of a set of cytosolic proteins or by motor proteins that attach membranes to the cytoskeleton.33 In addition to membrane deformations, the formation of endosomal compartments requires a series of fission and fusion events along the endosomal routes in which membrane and luminal components are exchanged and sorted. All of these events are initiated by regulatory small GTPases in the Rab and Arf family, which attach to the membrane and recruit a series of effector proteins34,35 (Figure 2).

Regulation of endosomal trafficking. Major regulatory proteins that shape endosomal and secretory pathway membranes and determine dynamics the extent of membrane flow. Endosomal compartments are indicated in white, and secretory pathway compartments are indicated in orange.
Scission of endocytic carriers from the plasma membrane and their fusion into/with early endosomes, including pre-early endosomal events and rapid recycling events, is regulated by Arf1 and Arf6 GTPases.23,24 The binding of Rab5 to early endosomes is essential for their maturation, including fusion and microtubule attachment, whereas the binding of Rab4 and Rab35 creates recycling domains that enables the production and release of recycling vesicles and facilitates fast recycling. The attachment of Rab11 to early endosomes leads to the formation of tubular extensions that convert early endosomes into a network of recycling tubules (JRC) and facilitate slow recycling. The process of slow recycling is complex and involves the concomitant activation of Rab11, Rab22a and most likely Arf1 and Arf6 GTPases.25,36 Replacement of Rab5 on early endosomes with Rab7 generates endolysosomal domains that bud extensively into the lumen of endosomes to form enlarged vacuolar and multivesicular endosomes that mature into late endosomes. The attachment and activation of Rab7b on late endosomes results in the formation of an endosomal domain with the capacity to generate endocytic carriers that travel towards the TGN,37 whereas the attachment of Rab27b and Rab27a leads to the formation of secretory endolysosomes and late endosomal recycling and exocytosis.29,31,32 The remaining late endosomes with attached Rab7 fuse with lysosomes and create degradation organelles38 (Figure 2).
The recruitment of regulatory proteins onto endosomal membranes is determined by the biochemical composition of the membrane (i.e., lipid composition) and activity of guanine nucleotide exchange factors and GTPase-activating proteins. The attachment of activated regulatory proteins is followed by the replacement and activation of effector proteins that change the physiological properties of the membrane, including its capacity for deformation, motility, fusion/fission, internal budding, tubulation and vacuolization, among others.34,35 Similar principles operate in the secretory pathway (Figure 2).
Routes of cargo molecule trafficking
Based on the composition of the endosomal system and the complexity of the regulatory network, it is clear that the trafficking of cargo molecules depends on many steps along the endocytic pathway. Thus, the routes of cellular proteins are not uniform and should be explored for every protein molecules. Several examples of relatively well-characterized routes are outlined in Figure 3.

Endocytic routes of some well characterized cargo proteins. (a) Tf-TfR, transferrin receptor (TfR) with bound ligand transferrin (Tf), route of clathrin-dependent cargo molecules that recycle via the fast and slow early endosomal recycling route; (b) EGF–EGFR, epidermal growth factor receptor with bound ligand (EGF), route of clathrin-dependent receptors that follow the late endosomal route when associated with ligand (red route) or recycle via early endosomes when unassociated with ligand; (c) LDL–LDLR, low-density lipoprotein receptor with bound ligand (LDL), route of receptor molecules that traffic into late endosomes when associated with ligand (red route) or rapidly recycle in the actin-dependent area when dissociated from ligand (green route); (d) CD63 route, a representative late endosomal resident protein that cycles between late endosomes and the plasma membrane; (e) GPI (glycosylphosphatidyl inositol)-anchored proteins (GPI-APs) follow a different route when associated with lipid rafts (rGPI-APs, red route) than those that reside in a lipid-disordered membrane environment (nrGPI-APs, green route); (f) the route followed by CD44 is an example of proteins that rapidly cycle in the cortical area of the cell (actin-dependent). Orange area, actin-dependent area; blue area, microtubule-dependent area.
Some cell surface receptors that contain specific motifs in their cytoplasmic domain that can recruit the AP-2 complex are constitutively taken up into clathrin-coated endocytic carriers that facilitate their rapid endocytic uptake.39 An example of this type of receptor is the TfR, which cycles in the early endosomal and juxtanuclear recycling route (Figure 3a). The route of some receptors, such EGFR, depends on ligand binding (Figure 3b). Unoccupied receptors are constitutively internalized via a clathrin-independent mechanism and mainly recycled back to the cell surface from early endosomes, the juxtanuclear recycling compartment or late endosomes40 (Figure 3b, green route). Ligand-associated EGFR is rapidly endocytosed by clathrin-dependent carriers that fuse with early endosomes and mature into late endosomes in which EGF/EGFR are trapped in intraluminal vesicles that are destined for degradation after fusion with lysosomes40 (Figure 3b, red route). Lysosomal targeting and degradation of EGF/EGFR is associated with ubiquitination of the receptor.41
After binding of lipoproteins at the cell surface, the LDL receptor is rapidly internalized via clathrin-dependent endocytosis, dissociated in pre-early endosomes and recycled back to the cell surface42 (Figure 3c, green route). Released lipoproteins (LDL) continue trafficking in early endosomes towards late endosomes, which contain the cellular machinery for the extraction of cholesterol and its routing towards various intracellular destinations43 (Figure 3c, red route).
Late endosomal resident proteins, such as CD63, Lamp1 and CD83, contain a tyrosine-based lysosomal-targeting motif44 that targets them and is retained in late endosomes (Figure 3d). Upon fusion of a subset of late endosomes with the plasma membrane (late endosomal recycling route), the recycled proteins are rapidly internalized via clathrin-dependent endocytosis into early endosomes and routed toward late endosomes.30
The trafficking route of membrane proteins that are attached to the plasma membrane by the GPI moiety (GPI-AP) depends on their localization in the membrane. When they partition into a lipid-disordered membrane microenvironment, they are endocytosed by the clathrin-independent pathway into GEECs, routed into the juxtanuclear recycling compartment and recycled back to the plasma membrane45 (Figure 3e, green route). When they partition into a lipid-organized membrane microenvironment (lipid-rafts), they are rerouted from early endosomes into late endosomes and returned to the plasma membrane via the late endosomal recycling route46 (Figure 3e, red route).
Some clathrin-independent membrane proteins, such as CD44, CD98 and CD147 (Figure 3f), are sorted by Hook1 protein and rapidly routed into recycling tubules, avoiding the classical early and late endosomal routes.16 This process of continuous recycling results in their prolonged lifetime on the cell surface.
Endocytic trafficking of antigen-presenting molecules
The trafficking route of MHC proteins is the best-characterized route of clathrin-independent cargo molecules. Fully conformed MHC I proteins are constitutively endocytosed into Arf6-associated carriers that fuse with TfR-loaded early endosomes.26,47 During trafficking along the early endosomal route, MHC I proteins are sorted into recycling domains and found in tubular early endosomes and the juxtanuclear recycling compartment (Figure 4a). In contrast to TfR, many endocytosed fully conformed MHC I proteins (approximately 40%) are directed into late endosomes and the degradation route.48,49 However, endosomal trafficking of MHC I proteins is more complex because MHC I molecules also can be displayed at the cell surface without antigenic peptide (empty MHC I, open MHC I conformers). In contrast to complete MHC I, they partition into distinct membrane microenvironments already present in the plasma membrane, endocytose into distinct endocytic carriers and are excluded from the early endosomal recycling domains (Figure 4b).48,49 Although the majority of empty MHC I molecules follow the degradation route, a portion of them segregate into a specific subset of late endosomes and undergo recycling via the late endosomal recycling route (Mahmutefendi_ et al., 2014, unpublished data).50 Thus, a detailed knowledge of the endocytic routes of both full and empty MHC I is essential to understand both antigen presentation mechanisms based on exogenous peptide loading and virus immune evasion strategies.

Endosomal routes of antigen-presenting molecules. (a) Endosomal route of fully conformed peptide-loaded major histocompatibility class I (full MHC I) and (b) peptide-empty MHC I molecules (empty MHC I) that sample peptide antigens in the cytosol and the endosomal compartments. (c) Nascent MHC II molecules associated with the invariant chain (Ii–MHC II) are directed into a late endosome-derived compartment (MIIC) where they sample peptide antigens (red route). Peptide-loaded MHC II molecules (pMHC II) rapidly recycle back to the cell surface after clathrin-independent endocytosis (green route). Endosomal trafficking of CD1a molecules (d) that follow the early endosomal recycling route as MHC I molecules, CD1b and CD1d molecules (e) are routed into a late endosome-derived antigen-presenting compartment (MIIC) from which they recycle back to the plasma membrane, and CD1c molecules (f) are equally distributed into the early and late endosomal route and recycled to the cell surface.
MHC II molecules have two distinct routes. Nascent MHC II molecules associate with the invariant chain (Ii) in the endoplasmic reticulum (ER), which inhibits peptide binding. Most, if not all, Ii–MHC II complexes travel via TGN to the cell surface and are rapidly endocytosed by clathrin-dependent carriers.51 Internalized Ii–MHC II are then delivered into a lysosome-like antigen-processing compartment (MHC II compartment; MIIC) where Ii is degraded and replaced with antigenic peptide. Peptide-loaded MHC II molecules (pMHC II) are then moved to the cell surface to display antigenic peptides to CD4 T lymphocytes (Figure 4c, red route).51 In contrast to Ii–MHC II, peptide-loaded MHC II that reach the cell surface are endocytosed by clathrin-independent endocytic carriers, rapidly internalized into early endosomes and recycled back to the cell surface (Figure 4c, green route).51 pMHC II internalize and reach a steady state ratio (80% at the surface, 20% intracellular) within 5 min, which indicates that cells possess efficient recycling machinery to maintain this distribution. pMHC II were found in Arf6+, Rab35+ and EHD1+ elongated tubules emanating from the plasma membrane.51
CD1 molecules, antigen-presenting molecules that are structurally related to MHC I, follow a route similar to MHC I proteins (Figure 4d–f). CD1a molecules traffic mainly through early endosomes and are recycled as MHC I proteins (Figure 4d), whereas CD1c molecules are equally distributed in early and late endosomes and recycled back to the cell surface also via the late endosomal recycling route (Figure 4f).52,53,54 In contrast, CD1d and CD1b are sorted mainly into late endosomes, into the same compartment as MHC II molecules (MIIC), and returned to the cell surface via the late endosomal recycling route (Figure 4e).54
Immune evasion activities of CMVs by perturbation of the endosomal system
There is increasing evidence that all members of the herpesvirus family can modulate some or all effector functions of the adaptive immune response. This modulation is based on the downregulation of cell surface molecules that participate in immune recognition, both on target and on effector cells. Mechanisms that are based on disruption of the cell surface egress from the secretory pathway of newly synthesized receptors or ligands are well understood, including responsible viral gene products.55,56 However, little is known about mechanisms that are based on the perturbation of endosomal trafficking of receptors and ligands that participate in immune recognition. This lack of information may be explained by a poor understanding of basic endosomal trafficking and endosomal perturbations in infected cells. Thus, in the following section, we provide an overview of current knowledge on the perturbation of the endosomal system and endosomal trafficking in CMV-infected cells. Unfortunately, the available data are insufficient to create an overall map of immune evasion events in the endosomal system. In general, viruses can downmodulate cell surface expression by altering the endocytic properties of a plasma membrane protein or by affecting the endosomal maturation program and thereby disrupting the physiological route of a given protein.
The host immune response to CMVs involves both innate and adaptive arms of the immune system. For efficient development of the adaptive immune response to CMVs, it is essential to prime CD8+ and CD4+ T cells.57 Efficient priming is based on the presentation of viral proteins by MHC I and MHC II molecules at the cell surface of either infected cells or antigen-presenting cells, killing the infected cells or initiating a specific immune response, respectively. Thus, processing of viral antigens in the MHC I and MHC II presentation pathway and recognition of MHC proteins are decisive targets for CMVs to evade effective clearance from the host.
MHC I molecules sample endogenous viral antigens that are processed in the cytosol using a sophisticated mechanism involving proteasomal degradation and peptide loading onto nascent MHC I proteins.58 Peptide-loaded MHC I proteins are displayed at the cell surface and monitored by TCR/CD3 complexes on CD8+ T lymphocytes. Thus, interference with peptide loading and trafficking of MHC I proteins to the cell surface is an efficient immune evasion strategy that evolved in CMVs (Figure 5). Additionally, cell surface MHC I proteins are constitutively endocytosed into endosomal compartments and recycled back to the cell surface from various points of the endosomal system23,49,59 (Figure 4). During endosomal trafficking, MHC I proteins are exposed to the acidified endosomal environment and mixture of internalized cargo molecules, including viral peptides,48 which also allows peptide sampling in the endosomal system and presentation of exogenous antigens (known as cross-presentation).60,61 Therefore, interference with the endosomal trafficking of MHC I proteins and inactivation of the antigen-presenting capacity of the endosomal environment is of particular interest for CMVs (Figure 5).

Major effector functions in the immune response to herpesviruses and immunoevasins of cytomegaloviruses that act either in the secretory or the endosomal pathway. The secretory pathway route is illustrated in orange and the endosomal pathway route in blue. Known immunoevasins that act in the secretory pathway and the endosomal pathway are listed in the boxes. Not known, indicates that there is evidence for CMV activity, but the viral gene product is not known. Not determined, indicates that there is no evidence for CMV activity. CMV, cytomegalovirus.
MHC II molecules are expressed on specialized cells (B lymphocytes, dendritic cells, monocytes/macrophages) known as antigen-presenting cells, which are essential for the development of adoptive immune response.62,63 MHC II molecules sample exogenously derived peptide antigens in a specialized region of the endosomal system of these cells (MIIC).62,63 Peptide-loaded MHC II proteins are exposed at the cell surface and constitutively cycle in the endosomal system (Figure 4), including passage through the acidic gradient of endosomal compartments and exposure to exogenous peptides obtained at the cell surface.64 Therefore, for MHC I, remodeling of MHC II trafficking in antigen-presenting cells or perturbation of the antigen-presenting compartment is of particular interest for CMVs to disrupt the development of the specific immune response (Figure 5).
NK cells are critical for defense against CMVs during the early stages of host infection and prior to the development of an effective adaptive immune response.3,65 They recognize the altered cell surface of infected cells and provide protection by releasing interferon gamma or by direct lysis of infected cells. Their activity is controlled by a balance of activating (i.e., human MICs, ULBPs, RAET1s and murine MULT-1, H60, RAE-1) and inhibitory (i.e., MHC I) signals that are received from the cell surface of the infected cell3,4,65,66 (Figure 5). Thus, modulation of the cell surface expression of activating or inhibitory molecules is of particular interest for CMVs to escape recognition by NK cells.4,65,66 This modulation can be achieved by reducing their egress to the cell surface from the secretory pathway or by rerouting their endosomal trafficking and redirecting them from the cell surface into the cell interior (Figure 5).
Recent studies indicate that NKT cells, a subset of T cells that co-express T-cell receptors and receptors of NK cells, are important during the early stages of the immune response to various pathogens, including herpesviruses.67 NKT cells can be activated by endogenous and exogenous antigenic lipid and glycolipid ligands exposed at the cell surface of infected cells by non-polymorphic MHC I-like proteins from the CD1 family (Figure 5).68 CD1a samples lipid antigens in the early recycling endosomes and CD1b samples the late endocytic compartment, whereas Cd1c samples the entire endosomal system in antigen-presenting cells.52 CD1d samples the endosomal system of all cells and thereby is crucial for early surveillance by NKT cells. Thus, a decrease in the cell surface expression of CD1 molecules by blocking their synthesis, export to the cell surface or by modifying their endosomal trafficking will modulate their sampling activity and activation of NKT cells (Figure 5).
Additionally, for those herpesviruses with tropism to immune effector cells, it is of interest either to reduce the overall expression or to perturb the endosomal trafficking of immune recognition molecules (i.e., TCR/CD3, CD4 molecule, costimulatory molecules, adhesion molecules) and thereby, reduce their cell surface expression. The spectrum of virus evasion targets by remodeling of endocytic trafficking is also confined by the cell tropism.
Murine CMVs (MCMV) remodels endosomal maturation in the early phase of infection, relocating cell surface MHC I into an endosomal retention compartment
MCMV encodes several immunoevasion proteins (immunoevasins) that associate with nascent MHC I molecules in the ER and cooperatively prevent supply to the cell surface of functional MHC I proteins (Figure 5). Protein m152 retains these proteins in the ER-ERGIC route.69,70,71 Protein m04 forms complexes with MHC I and escorts it to the cell surface,71,72 whereas protein m06 redirects it into lysosomes for degradation.71,73 The net result of this cooperative activity is the loss of MCMV peptide-loaded MHC I proteins from the cell surface (downregulation of MHC I after prolonged activity of immunoevasins) and thus, the prevention of presentation to CTL.71,74 Experiments with mutant viruses containing a deletion suggest that no other immunoevasins in the MCMV genome are required for MHC I downregulation.74 However, in MCMV-infected fibroblasts, almost all of the MHC I proteins are rapidly cleared from the cell surface at 5–7 h after infection75 (Figure 6). This loss occurs much earlier than expected due to the constitutive uptake of cell surface resident MHC I proteins under inhibited supply conditions by the cooperative activity of MCMV immunoevasins. Namely, the half-life of cell surface resident MHC I proteins is longer than 10 h under conditions of prevented egress (Figure 6), and constitutive internalization occurs at a rate of 4%–10% per hour.48,49,76 Thus, in the absence of additional MHC I immunoevasins, the rapid removal of MHC I from the cell surface in the early phase of MCMV infection can be explained by perturbation of the endosomal route.

Rapid downregulation of cell surface MHC I during the early phase of infection with MCMV. Cell surface expression of H2-Kd molecules was determined by indirect immunofluorescence and flow cytometry using the monoclonal antibody MA-215 with murine embryonic fibroblasts at various times after treatment with BFA (10 µg/ml) and after MCMV infection (infection with w.t. MCMV, multiplicity of infection of 20). BFA, Brefeldin A; MCMV, murine cytomegalovirus.
We have recently shown that MCMV perturbs endosomal trafficking very early during the infection by acting on distal parts of the early endosomal route and generating the early phase retention compartment.75,77 This compartment has the characteristics of sorting endosomes at the terminal stage of their maturation. It contains markers of early endosomes (EEA1 and Rab5) but not markers of late endosomes (i.e., LBPA, GM1). It also retains incoming cargo molecules that travel through both the recycling (i.e., TfR, MHC I) and endolysosomal (EGFR, Lamp1) routes.75 Clathrin-dependent cargo protein (TfR), which is known to be exhaustively recycled after endocytosis, is rapidly displaced from the cell surface of MCMV-infected cells as in uninfected cells (Figure 7a, middle) and accumulates in the perinuclear area (Figure 7a, right), from which it cannot leave (Figure 7a, middle) due to recycling inhibition (Figure 7a, left).75 Clathrin-independent cargo proteins (MHC I) are constitutively internalized in MCMV-infected fibroblasts at a higher rate than in uninfected cells (Figure 7b, middle) and accumulate in the perinuclear retention compartment (Figure 7b, right) together with internalized TfR.75 Their internalization rate is much higher in MCMV-infected fibroblasts (Figure 7b, left) due to the decreased recycling rate,75 suggesting that MCMV infection inhibits recycling from juxtanuclear endosomes. Furthermore, our recent analysis of the CD44 route in MCMV-infected cells (Figure 7c, left) suggests that MCMV infection also perturbs proximal steps in endosomal recycling. The internalization rate of CD44 also increases in MCMV-infected cells (Figure 7c, middle), and a significant fraction of CD44 is displaced in EEA1+ perinuclear endosomes as soon as 4 hrs post-infection (Figure 7c, right). Thus, it seems that MCMV infection extensively perturbs early endosomal trafficking during the early phase of infection, including the formation of recycling endosomal carriers and maturation of early endosomes into late endosomes, which results in the development of the perinuclear retention compartment. This early endosomal retention is associated with a reduction of the intracellular content of Rab11 and Rab7,75 small GTPases that are essential for the formation of the recycling and endolysosomal domains of early endosomes, respectively.34,35 The levels of cell surface proteins, such as MHC I molecules, depend on the recycling efficiency,48,49 and inhibition of recycling may result in rapid cell surface loss and thereby, indirectly enable evasion of immune recognition by effector T cells.

Endosomal routes of clathrin-dependent and clathrin-independent cargo proteins in MCMV-infected cells during the early phase of infection. (a) The endosomal route of the TfR in MCMV-infected MEFs at 6 h p.i. (left). The cell surface levels of TfR (middle) were determined by flow cytometry using the mAb R17 217.1.3 (ATCC TIB-219; anti-rat IgG FITC as secondary antibody) after pulse labeling (60 min at 4 °C) and 0–60 min internalization at 37 °C. Confocal images (right) of the intracellular distribution of internalized TfR after cell surface labeling at 4 °C with the mAb R17 and 0–60 min internalization at 37 °C. A short acid wash (pH 2.0) was performed to remove un-internalized mAb–TfR complexes from the cell surface before staining with anti-rat IgG-AF555. (b) The endosomal route of MHC I molecules in MCMV-infected MEFs (left). The internalization profile of H2-Kd molecules (middle) in uninfected and 6 h-infected MEFs determined by flow cytometry after labeling at 4 °C (60 min) with mAb MA-21579 and internalization at 37 °C for 0–60 min.48 Anti-mouse IgG-FITC was used as a secondary antibody. Confocal images (right) of the cell surface and intracellular Kd of uninfected and MCMV-infected MEFs (4, 6 and 8 h p.i.). The cell surface and intracellular Kd were stained in intact and Triton X-100-permeabilized cells with mAb MA-215 and anti-mouse IgG2a AF555 (red fluorescence). Infection was verified by simultaneous staining of the MCMV IE1 using mAb CRO-10177 and anti-mouse IgG1 AF488 (green fluorescence). (c) The endosomal route of CD44 protein in MCMV-infected cells at 6 h p.i. (left). The internalization profile of CD44 (middle) in uninfected and 6-h-infected MEFs determined by flow cytometry after labeling at 4 °C (60 min) with mAb IM7 and internalization at 37 °C for 0–60 min. Anti-rat IgG-FITC was used as the secondary antibody. Confocal images of the intracellular distribution of CD44 in MCMV-infected cells at 2 and 4 h p.i. (right). Fixed and permeabilized infected cells were simultaneously stained with mAb IM7 (CD44) and mAb against EEA1 (Zymed Laboratories, San Francisco, CA, USA) followed by the secondary reagents anti-rat IgG AF555 (red fluorescence) and anti-chicken Ig AF488 (green fluorescence), respectively. EEA1, early endosomal antigen 1; IE1, immediate early 1 protein; mAb, monoclonal antibody; MCMV, murine cytomegalovirus; MEF, murine embryonic fibroblast; p.i., post-infection; TfR, transferrin receptor.
In addition to MHC I, remodeling of endosomal trafficking during the early phase of MCMV infection can contribute to other immunoevasion activities of MCMV, especially the downmodulation of NKG2D ligands. Three of them (RAE-1ε, MULT-1 and H60) are downregulated from the cell surface via a mechanism that involves the inhibition of their endosomal recycling.4,78 Thus, additional studies on the endosomal trafficking routes of other molecules that participate in immune recognition during MCMV infection are required, including NKG2D ligands, to better understand the physiology of MCMV immunoevasion.
Human cytomegalovirus (HCMV) extensively remodels the endosomal system
Remodeling of the endosomal system in HCMV-infected cells is associated with the development of the cytoplasmic ‘assembly compartment'.80,81 This compartment is a complex juxtanuclear structure that develops by reorganization of cellular organelles at sites of viral tegument, envelope and nonstructural protein accumulation to create an environment for the final tegumentation, envelopment and preparation for shedding of infectious virions.80,81,82,83,84,85,86 Recent data indicate that the assembly complex develops by dramatic rearrangement of the secretory and endocytic organelles in a way that endosomal membranes and TGN-derived vesicles are recruited to the cell center and represent the ‘virus factory', while the Golgi and lysosomes are excluded and surround the site of assembly.83,84,87,88 In addition to viral tegument proteins80,81,82,83,85,86 and envelope glycoproteins,80,81,82 this compartment also accumulates TGN markers (i.e., TGN46),80,81,83,84,87 redistributed early endosomal markers (i.e., EEA1, annexin I),84,87 and cargo molecules that travel via the early endosomal route (i.e., M6PR, HRS)87 and the recycling route (i.e., TfR,).87 It does not contain lysosomal markers (i.e., Lamp1),87 but contains CD6382,83,87,88 and marker of MVBs, which also suggests the recruitment of MVB membranes to the site of virion assembly. In cell culture, the assembly complex develops slowly over 3–4 days of remodeling before the first virions are released.81,83,84,86 The remodeling is a stepwise recruitment and expansion of existing vesicular membranes and related regulatory components, which is associated with the perturbation of trafficking routes, both in the secretory and endosomal pathways.
The mechanism(s) of membranous organelle remodeling remains poorly understood. It involves extensive rearrangements of the cytoskeleton and modulation of the expression of regulatory and effector proteins that shape the endosomal and vesicular system. It has been shown at later times postinfection that HCMV upmodulates 81 genes and downmodulates 132 cellular genes that affect functions involved in vesicular trafficking.89 In response to infection, transcription of the arf1 and arf6 genes and 13 members of the rab family genes (rab1a, -1b, -2a, -3a, -6a, -7, -13, -18, -21, -23, -31, -34 and -40b) is downmodulated; in contrast, seven rab family genes (ray/rab1C and rab2L, -8, -20, -27a, -32 and -38) were upmodulated.89 Thus, a proper understanding of the biogenesis of the endosomal system will help reconstruct the events that lead to remodeling of the endosomal and secretory system during HCMV infection. It is still being debated whether HCMV infection expands existing compartments or generates a novel hybrid compartment with both endosomal and TGN properties. Recent data suggest that HCMV may expand transport vesicles between endosomes, TGN87 and late endosomal-derived MVBs.80,83,90 Consistent with these findings, it was shown in HCMV-infected cells that there a threefold increase in the expression of Rab27a,91 a small GTPase that regulates secretion via late endosome-derived lysosomal-related organelles.29 The association of Rab27a with the assembly complex suggests that HCMV exploits the machinery of endolysosomal secretion for its assembly. Upregulation of Rab27a is also associated with heterotypic fusion of Rab11+ membranes (early endosomal membranes of the recycling route) with Rab27b+ multivesicular bodies (late endosomal subset that generate secretory endosomal organelles).29,31,91 The process can be initiated by viral glycoproteins that recruit endosomal effector proteins, i.e., HCMV gM forms a dimer with gN and interacts with the Rab11 effector protein FIP4 (family of interacting protein 4) to form an intracellular ternary complex in the juxtanuclear recycling compartment.92 In addition, it has been shown recently that HCMV encodes multiple microRNAs (miRNAs US5-1, US5-2 and UL112-1) to target various components that shape the vesicular pathway (i.e., Rab5c, Rab11a and Cdc42) and thereby, coordinately reorganize secretory pathway and perhaps endosomal organelles.93
Present data restrict the effect of HCMV on MHC I expression to the earliest stages of MHC I assembly and trafficking in the secretory pathway
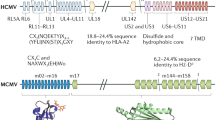
HCMV encodes multiple genes to evade MHC I antigen presentation and immune recognition by CD8+ cytotoxic T lymphocytes (Figure 5). Similarly to MCMV, HCMV infection eliminates cell surface MHC I expression by preventing the supply to the cell surface of newly formed peptide-loaded MHC I proteins by affecting several steps in MHC I biogenesis and transport (Figure 8). The product of the HCMV US6 gene attacks the TAP (transporter associated with antigen presentation) complex and blocks peptide loading onto MHC I molecules in the ER,94 while products of the US3 and US11 genes dislocate newly synthesized MHC I heavy chains from the ER into the cytosol for degradation.95,96 MHC I proteins that successfully assemble and escape destruction are captured by the US3 gene product and retained in the ER.97,98 The cooperative activity of HCMV immunoevasins would result in a gradual loss of MHC I from the cell surface with kinetics that correspond to their constitutive internalization rate (5%–10% per hour49). These kinetics occur in cultured fibroblasts in which HCMV replication is slow.99 Although there is no evidence that perturbation of the endosomal route contributes to the clearance of MHC I proteins from the cell surface, HCMV appears to perturb the same endosomal routes of other cellular proteins as MCMV. Consistent with these findings, recent data indicate that HCMV inhibits TfR recycling86 and rapidly downregulates EGFR expression.100

Known sites of herpesvirus activity in the intracellular trafficking of immune recognition molecules. Members of the herpesvirus family block the exit of newly synthesized immune recognition molecules from the biosynthetic pathway (green), the return of internalized molecules via early endosomal recycling (orange) and late endosomal recycling (grey), or the rerouting from the early endosomal recycling route to the late endosomal route and lysosomal degradation (blue).
In conclusion, in cells in which the replication cycle of HCMV is slow, the effect of HCMV immediate early gene products on MHC I in the secretory pathway is sufficient to ensure MHC I eradication from the cell surface. However, in cells with a much faster replication cycle, endosomal remodeling should contribute significantly as an immunoevasion mechanism. This phenomenon has been shown for murine75 and rat CMV infection,101,102 but should also be considered for HCMV infection in vivo. The reputation of HCMV as a slowly replicating virus is based on the development of a cytopathic effect in cell culture. However, studies in vivo suggest that the replication of HCMV in vivo is highly dynamic, especially in immunocompromised patients.103
HCMV inhibits late endosomal recycling and prevents egress of peptide-loaded MHC II proteins from the peptide-loading compartment during productive and latent infection
In addition to MHC I trafficking, HCMV has evolved mechanism(s) that alter MHC II expression. These mechanisms are based on remodeling of their endosomal route and do not relate to known mechanisms that downregulate MHC I in the secretory pathway.
HCMV infection reduces cell surface expression of MHC II in dendritic cells,104,105 monocytes/macrophages106,107 and professional antigen presenting cells (mature Langerhans cells).108 These cells are major targets for HCMV infection, latency and reactivation. The mechanism of MHC II cell surface down-regulation was successfully characterized in transfected astrocytoma cells that constitutively express MHC II.109 In HCMV-infected astrocytoma cells109 and HCMV-infected mature Langerhans cells,108 the early stages of endosomal transport (including recycling), entry into a peptide-loading compartment (MHC II) and assembly and peptide loading of MHC II are unaltered, but the latest stages of their transport are blocked. Peptide-loaded MHC II is retained in the perinuclear area and sequestered in an enlarged peptide-loading compartment with defective trafficking of MHC II-loaded vesicles toward the periphery.108,109 In addition to MHC II, HCMV is also sequestered in the same location as the costimulatory molecules CD80, CD83 and CD86,108,110,111,112 suggesting that HCMV attacks the late endosomal recycling route. All of these effects are not induced by the MHC I immunoevasins that localize in US1–US11 region.109 In addition to endosomal perturbation, the immunoevasion potential of HCMV is further extended to cytoskeletal remodeling. In HCMV-infected mature Langerhans cells, the loss of peptide-loaded MHC II from the cell surface is associated with the loss of cytoskeletal extensions that provide support for dendrites of Langerhans cells,108 which reduces their ability to cluster with T cells and decreases their capacity to stimulate T-cell proliferation.113,114
Immunoevasion by endosomal perturbation is even more attractive in latently infected cells in which the expression of HCMV genes is restricted to CMV latency-associated transcripts, which are encoded by the immediate early (ie1/ie2) region of the viral genome.115 These transcripts are expressed in natural and experimental latent infection. In experimental latent infection of granulocyte-macrophage progenitor cells in culture that do not express productive viral replication gene products, the cell surface level of MHC II is markedly reduced, synthesis and processing occur normally, and MHC II proteins together with HLA-DM are redistributed and retained within punctate cytoplasmic vesicles.116 This reduction of cell surface MHC II is independent of all known immunomodulatory functions (i.e., in the US2–US11 region mutant, MHC II is downregulated and is not expressed during latent infection).116 Thus, it appears that HCMV also perturbs the endosomal trafficking of MHC II during latent infection. The mechanism of perturbation is unclear. However, the observation that 50% of latently infected cells down-regulate MHC II and only 2%-3% contain of them contain latency associated transcripts suggests that that down-regulatioion information may be distributed in cell culture, perhaps by miRNA.
HCMV evades NK cell-mediated lysis by a variety of different mechanisms that are not associated with endosomal perturbation
HCMV has evolved a variety of different mechanisms to evade NK cell activation66 that are based either on the expression of molecules that engage inhibitory NK receptors or on the down-modulation of ligands that engage activating NK receptors (Figure 5). Surface expression of HLA-E enabled by HCMV UL40, virus-encoded MHC I homologue UL18, tegument protein pp65, and glycoprotein UL141 are examples of HCMV-encoded functions that engage inhibitory receptors.66 HCMV also downregulates ligands that activate NKG2D (MICs, ULBPs, RAET1s),117 either by downregulating their transcription by virus-encoded microRNAs (MICB by miRUL112)118 or by retaining them in the ER/cis-Golgi (MICB, ULBP1, ULBP2 and ULBP6 by UL16)119 or in the Golgi (MICA and ULBP3 by UL142).120
Given that HCMV-infected cells are extremely resistant to NK cell lysis, it can be anticipated that the number of HCMV genes possessing NK cell-activating ligands, including HCMV-modulated cellular functions, will increase with further research. For example, two additional HCMV genes, US18 and US20, have been recently identified to promote downregulation NK cell-activating ligands by lysosomal degradation121 (Figure 5). This observation is the only association, to our knowledge, of a NK immunosubversive function associated with endocytic trafficking. A further understanding of a HCMV immunosubversion mechanism via down-modulation of NK cell-activating ligands, or at least MICs, will require answering the question of how HCMV deals with sequence polymorphisms of MICs (80 alleles of MICA and 33 alleles of MICB). A mechanism based on modulation of the endosomal route has been proposed recently by Agüera-González et al.122 The short-term residence of MCIB at the cell surface of the HCMV-infected cell may result in its redistribution into a compartment that shares features with late endosomes/lysosomes, but is functionally and morphologically distinct.122 Thus, remodeling of endocytic trafficking in concert with known activities of immunoevasins in the secretory pathway should be considered.
Immune evasion potential of endosomal perturbation during infection with other herpesviruses
Other herpesviruses also have evolved a variety of mechanisms to disrupt the cell surface display of immune recognition molecules. In addition to early identified effects on their biogenesis, substantial evidence indicates that herpesviruses also target endosomal trafficking. The sites of immunoevasion activities of human herpesviruses are indicated in Figure 8.
Several herpesviruses modulate the earliest steps of MHC I biogenesis and trafficking in the secretory pathway. Herpes simplex virus 1 and 2 (HSV-1 and HSV-2), members of the alpha-herpesvirus subfamily, encode the ICP47 protein that inhibits peptide loading of MHC I proteins in the ER and thereby prevent the export of MHC I to the cell surface.123,124 Similar tools also evolved in the Epstein–Barr virus (EBV), Kaposi's sarcoma-associated virus and murine herpesvirus 68, which are members of the gamma-herpesvirus subfamily. These viruses encode proteins (EBNA1 of EBV,125 LANA1 of KSV126 and ORF73 of murine herpesvirus 68127) that suppress peptide loading of MHC I and prevent the exit of MHC I proteins from the ER. Varicella-zoster virus, another member of the alpha-herpesvirus subfamily, acts a bit later in the secretory route of MHC I. It encodes an Us3 homolog that arrests MHC I in the cis/medial-Golgi and retains it in the perinuclear area.128
The next step in MHC I trafficking along the secretory pathway chosen by herpesviruses is their rerouting in the trans-Golgi towards rearranged late endosomal compartments. In addition to murine CMV, human herpesvirus 6 (HHV-6) and HHV-7, beta-herpesviruses, which are closely related to CMV that infects CD4 T lymphocytes, encode the U21 gene. The product of U21 associates with properly folded peptide-loaded classical and non-classical MHC I proteins in the ER and reroutes them from the Golgi into the endolysosomal compartment, thereby effectively removing them from the cell surface.129,130,131 The U21 protein also binds to NK cell-activating ligands (ULBP1, MICA and MICB ligands for NKG2D),131 but it does not bind to the NKT ligand CD1d.130 U21-expressing cells sequester MHC I and ULBP1 in a perinuclear location in late endosomes and facilitate their rapid degradation.130,131 Unlike MCMV m06, U21 does not have a di-leucine motif to reroute trafficking towards late endosomes, which suggests that U21 uses an unidentified cellular protein(s) that contains lysosomal targeting information in its cytoplasmic tail and binds to the luminal domain of MHC I and NKG2D ligands.130
In addition to the rerouting activity of U21 in the Golgi, HHV-6 and HHV-7 encode protein U24, which functions in the endosomal route. The activity of U24 does not affect MHC I trafficking but inhibits constitutive recycling of the T-cell receptor complex (TCR-CD3) in infected CD4 T lymphocytes and retains them in Rab4+ and Rab5+ early endosomes.132 Given that U24 also inhibits the recycling of TfR through the PPXY motif near the amino terminus; it appears that U24-mediated downregulation of TCR-CD3 results from a general block in the recycling route used by TCR-CD3 and TfR, but not in the recycling route used by MHC I.132
Disruption of endosomal recycling or rerouting in early endosomes and exclusion from the recycling route is a preferable target not only for beta-herpesviruses but also for other herpesviruses. Endosomal recycling is a key cellular mechanism to control the number of molecules at the cell surface. In the case of antigen-presenting molecules, recycling is a crucial step, especially for cross-presentation. Perturbation of the recycling route of antigen-presenting and immune recognition molecules, either directly or indirectly, is therefore a logical target for herpesviruses (Figure 8).
Studies of HSV-1-infected antigen-presenting cells have demonstrated that HSV-1 disrupts the endosomal recycling machinery, which strongly reduces the cell surface expression of CD1b and CD1d molecules.133,134 Constitutively internalized CD1d molecules are redirected from the recycling route and accumulate intracellularly with a late endosome phenotype where they colocalize with viral proteins.133,134 Thus, although there is no experimental evidence that the same mechanism affects MHC I trafficking in HSV-infected cells, there is a strong indication that the endosomal route of MHC I is affected in HSV-1-infected cells. It has been shown that HSV-1 Us3 kinase activity is required for efficient cell surface downregulation of MHC I proteins in HSV-1-infected cells.135 Us3 does not phosphorylate MHC I and cannot downregulate MHC I expression when over-expressed alone, which suggests that it downregulates cell surface MHC I indirectly.135
Other members of the alpha-herpesvirus subfamily, equine herpesviruses (EHV-1 and EHV-4)136,137 and bovine herpesvirus 1,138 rapidly downregulate cell surface MHC I very early in infection by a mechanism that involves enhanced internalization, perhaps disrupting their recycling routes. Downregulation of cell surface MHC I in EHV-1- and EHV-4-infected cells is observed as early as four hours postinfection and is associated with the expression of pUL56, a product of ORF1; however, the mechanism is poorly characterized.137
The endosomal rerouting of cellular proteins that participate in immune recognition appears to be a dominant immune evasion strategy of gamma-herpesviruses. Kaposi's sarcoma-associated virus encodes ubiquitin ligases (kK3 and kK5, known as MIR1 and MIR2, respectively) that interact with transmembrane domains of cell surface MHC I proteins to catalyze their polyubiquitination and orchestrate their rapid removal from the cell surface.139,140 Polyubiquitination redirects constitutive endocytic uptake of MHC I proteins into the clathrin-dependent pathway, which accelerates their endocytic uptake,141 and excludes internalized MHC I proteins from the endosomal recycling routes. In early endosomes, ubiquitinylated MHC I proteins are sorted into endolysosomal domain, rerouted into late endosomes and from there, via the late endosomal sorting protein TSG-101, delivered into the lysosomal compartment for degradation.142 It has been shown that kK5 has a remarkable ability to target and downregulate not only MHC I but also many immunoreceptors, including ICAM-1,143 CD86,143 CD1d,144 MICA/B,145 AICL,145 PECAM,146 ALCAM147 and IFN receptors,148 and thereby, effectively prevent recognition by cells of the adaptive (MHC I and CD1d restricted NKT cells) and the innate (NKG2D- and NKp80-restricted NK cells) immune response. In addition to polyubiquitin-mediated endosomal rerouting to lysosomal degradation (i.e., MHC I139 and AICL145), some immunoreceptors are downregulated by perturbation of the endosomal system. For example, Cd1d144 and NKG2D ligand MICA145 are not degraded, but redistributed and retained in an enlarged intracellular compartment that lacks clathrin, EEA1, Vps-26, Lamp1 and TGN-46.145
EBV encodes a protein, BILF1, which associates with MHC I at the cell surface and targets it for lysosomal degradation.149 In contrast to Kaposi's sarcoma-associated virus kK5, BILF1 does not mediate MHC I downregulation by polyubiquitin-enhanced endocytosis and does not reroute them towards lysosomes.150 It seems that BILF1 cooperates with other EBV genes that generally perturb endosomal routes to reduce cell surface MHC I.150 This perturbation is associated with dislocation and intracellular retention of MHC I together with the costimulatory molecules CD80 (B7-1), CD83 and CD86 (B7-2) but not MHC II.151
Conclusions
Remodeling of the endosomal system is a hallmark of infection with herpesviruses, especially with CMVs. This remodeling is evident during the latest stages of CMV infection. In HCMV-infected cells, at least a part of the remodeling is associated with the development of the assembly compartment; however, very little is known about perturbations in earlier stages of infection. These perturbations might be associated with several important but still unresolved issues in CMV biology, such as cell tropism, productive infection and latency. A better understanding of the physiology of endosomal organelles and intracellular pathways will facilitate our knowledge of endosomal remodeling during CMV infection. Thus, it is important to determine the physiological route of many intracellular proteins to understand the physiology of the infected cell.
The same principles would apply to cellular proteins that contribute to immune recognition. The primary target for CMVs will be modulation of antigen presentation in the endosomal system, either by retrieval of MHC I loaded with viral peptides into endosomal compartments or redirection of MHC I trafficking and disruption of endosomal peptide loading. Thus, it is not surprising that CMVs target endosomal recycling because this recycling is essential for the maintenance of MHC I at the cell surface and for the mechanism of exogenous peptide loading. Still, the peptide-loading endosomal compartment has not been discovered, and the physiological role of MHC I proteins lacking peptide remains unknown. Hopefully, research on MHC I trafficking and exogenous MHC I peptide loading in infected cells will provide additional insight into the physiology of exogenous antigen presentation and immune recognition of infected cells.
Over the last decade, endocytosis research has revealed enormous complexity of the endosomal system and its functions. The complex molecular network that shapes the endosomal system ensures a high degree of plasticity, which makes interpretation of endosomal functions enormously complex, especially considering that many conclusions are based on long-term remodeling by siRNA, dominant-negative mutants or over-expression of individual molecular players. Thus, the physiological hierarchy of the molecular mechanisms has been difficult to establish. Remodeling by viruses, which is usually a rather fast process, may help to establish the patterns of molecular activities that shape physiological pathways in the endosomal system.
References
Roizman B . Herpesviridae. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 3rd ed. Philadelphia, PA: Lippincott-Raven Publishers, 1996: 2221–2230.
Lilley BN, Ploegh HL . Viral modulation of antigen presentation: manipulation of cellular targets in the ER and beyond. Immunol Rev 2005; 207: 126–144.
Lodoen MB, Lanier LL . Viral modulation of NK cell immunity. Nat Rev 2005; 3: 59–69.
Slavuljica I, Krmpotić A, Jonjić S . Manipulation of NKG2D ligands by cytomegaloviruses: impact on innate and adaptive immune response. Front Immunol 2011; 2: 85.
Terrazzini N, Kern F . Cell-mediated immunity to human CMV infection: a brief overview. F1000Prime Reports 2014; 6: 28.
Koenig JA, Edwardson JM . Endocytosis and recycling of G protein-coupled receptors. Trends Pharmacol Sci 1997; 18: 276–287.
Steinman RM, Mellman IS, Muller WA, Cohn ZA . Endocytosis and the recycling of plasma membrane. J Cell Biol 1983; 96: 1–27.
McMahon HT, Boucrot E . Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 2011; 12: 517–533.
Hansen CG, Nichols BJ . Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci 2009; 122: 1713–1721.
LeRoy C, Wrana JL . Clathrin- and nonclathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol 2005; 6: 112–26.
Gong Q, Huntsman C, Ma D . Clathrin-independent internalization and recycling. J Cell Mol Med 2008; 12: 126–144.
Kirkham M, Parton RG . Clathrin-independent endocytosis: new insights into caveolae and noncaveolar lipid raft carriers. Biochim Biophys Acta 2005; 1746: 349–63.
Sandvig K, Pust S, Skotland T, van Deurs B . Clathrin-independent endocytosis: mechanisms and function. Curr Opin Cell Biol 2011; 23: 413–420.
Mayor S, Pagano RE . Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 2007; 8: 603–612.
Massol RH, Larsen JE, Kirchhausen T . Possible role of deep tubular invaginations of the plasma membrane in MHC-I trafficking. Exp Cell Res 2005; 306: 142–149.
Maldonado-Báez L, Williamson C, Donaldson JG . Clathrin-independent endocytosis: A cargo-centric view. Exp Cell Res 2013; 319: 2759–2769.
Coskun Ü, Simons K . Membrane rafting: from apical sorting to phase segregation. FEBS Lett 2010; 584: 1685–1693.
Berditchevski F, Odintsova E . Tetraspanins as regulators of protein trafficking. Traffic 2007; 8: 1–8.
Vereb G, Szollosi J, Matko J, Nagy P, Farkas T, Vigh L et al. Dynamic, yet structured: the cell membrane three decades after the Singer–Nicolson model. Proc Natl Acad Sci USA 2003; 100: 8053–8058.
Swanson JA, Watts C . Macropinocytosis. Trends Cell Biol 1995; 5: 424–428.
Huotari J, Helenius A . Endosome maturation. EMBO J 2011; 30: 3481–3500.
Jovic M, Sharma M, Rahajeng J, Caplan S . The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol 2010; 25: 99–112.
Grant BD, Donaldson JG . Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 2009; 10: 597–608.
Hao M, Maxfield FR . Characterization of rapid membrane internalization and recycling. J Biol Chem 2000; 275: 15279–15286.
Weigert R, Yeung AC, Li J, Donaldson JG . Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell 2004; 15: 3758–3770.
Naslavsky N, Weigert R, Donaldson JG . Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol Biol Cell 2004; 15: 3542–3552.
Rink J, Ghigo E, Kalaidzidis Y, Zerial M . Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005; 122: 735–749.
Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, Mayran N et al. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J 2002, 21: 1289–1300.
Marks MS, Heijnen HFG, Raposo G . Lysosome-related organelles: unusual compartments become mainstream. Curr Opin Cell Biol 2013; 19: 495–505.
Braulke T, Bonifacino JS . Sorting of lysosomal proteins. Biochim Biophys Acta 2009; 1793: 605–614.
Laulagnier K, Schieber NL, Maritzen T, Haucke V, Parton RG, Gruenberg J . Role of AP1 and Gadkin in the traffic of secretory endo-lysosomes. Mol Biol Cell 2011; 22: 2068–2082.
Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 2010; 12: 19–30.
McMahon HT, Gallop JL . Membrane curvature and mechanisms of dynamic cell remodelling. Nature 2005; 438: 590–596.
D'Souza-Schorey C, Chavrier P . ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 2006; 7: 347–358.
Hutagalung AH, Novick PJ . Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 2011; 91: 119–149.
Donaldson JG, Williams DB . Intracellular assembly and trafficking of MHC class I molecules. Traffic 2009; 10: 1745–1752.
Progida C, Cogli L, Piro F, de Luca A, Bakke O, Bucci C . Rab7b controls trafficking from endosomes to the TGN. J Cell Sci 2010; 123: 1480–1491.
Vanlandingham PA, Ceresa BP . Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. Biol Chem 2009; 284: 12110–12124.
Benmerah A, Lamaze C . Clathrin-coated pits: Vive la difference? Traffic 2007; 8: 970–982
Sorkin A, Goh LK . Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res 2008; 314: 3093–3106.
Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A . Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell 2006; 21: 737–748.
Pompey S, Zhao Z, Luby-Phelps K, Michaely P . Quantitative fluorescence imaging reveals point of release for lipoproteins during LDLR-dependent uptake. J Lipid Res 2013; 54: 744–753.
van der Kant R, Fish A, Janssen L, Janssen H, Krom S, Ho N et al. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J Cell Sci 2013; 126: 3462–3474.
Berditchevski F, Odintsova E . Tetraspanins as regulators of protein trafficking. Traffic 2007; 8: 89–96.
Sabharanjak S, Sharma P, Parton RG, Mayor S . GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell 2002; 2: 411–423.
Fivaz M, Vilbois F, Thurnheer S, Pasquali C, Abrami L, Bickel PE et al. Differential sorting and fate of endocytosed GPI-anchored proteins. EMBO J 2002; 21: 3989–4000.
Naslavsky N, Weigert R, Donaldson JG . Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol Biol Cell 2003; 14: 417–431.
Mahmutefendić H, Blagojević G, Ilić Tomaš M, Kučić N, Lučin P . Segregation of open Major Histocompatibility class I conformers at the plasma membrane and during endosomal trafficking reveals conformation-based sorting in the endosomal system. Int J Biochem Cell Biol 2011; 43: 504–515.
Zagorac GB, Mahmutefendić H, Ilić Tomaš M, Kučić N, Le Bouteiller P, Lučin P . Early endosomal rerouting of Major Histocompatibility class I conformers. J Cell Physiol 2012; 227: 2953–64.
Mahmutefendić H, Zagorac GB, Tomaš MI, Groettrup M, Momburg F, Lučin P . Endosomal trafficking of open Major Histocompatibility Class I conformers—implications for presentation of endocytosed antigens. Mol Immunol 2013; 55: 149–52.
Walseng E, Bakke O, Roche PA . Major histocompatibility complex class II–peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem 2008; 283: 14717–14727.
Sugita M, van der Wel N, Rogers RA, Peters PJ, Brenner MB . CD1c molecules broadly survey the endocytic system. Proc Natl Acad Sci USA 2000, 97: 8445–8450.
Barral DC, Cavallari M, McCormick PJ, Garg S, Magee AI, Bonifacino JS et al. CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic 2008; 9: 1446–57.
Barral DC, Brenner MB . CD1 antigen presentation: how it works. Nat Rev Immunol 2007; 7: 929–941.
Hansen TH, Bouvier M . MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol 2009; 9: 503–513.
Alcami A, Koszinowski UH . Viral mechanisms of immune evasion. Immunol Today 2000; 21: 447.
Mitrović M, Arapović J, Jordan S, Fodil-Cornu N, Ebert S, Vidal SM et al. The NK cell response to mouse cytomegalovirus infection affects the level and kinetics of the early CD8+ T-cell response. J Virol 2012; 86: 2165–2175.
Pamer E, Cresswell P . Mechanisms of MHC class I-restricted antigen processing. Annu Rev Immunol 1998; 16: 323–358.
Jovanovic OA, Brown FD, Donaldson JG . An effector domain mutant of Arf6 implicates phospholipase D in endosomal membrane recycling. Mol Biol Cell 2006; 17: 327–335.
Bevan MJ . Cross-priming. Nat Immunol 2006; 7: 363–365.
Heath WR, Carbone FR . Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol 2001; 1: 126–134.
Neefjes J, Jongsma ML, Paul P, Bakke O . Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11: 823–836.
Rocha N, Neefjes J . MHC class II molecules on the move for successful antigen presentation. EMBO J 2008; 27: 1–5.
Dani A, Chaudhry A, Mukherjee P, Rajagopal D, Bhatia S, George A et al. The pathway for MHC II-mediated presentation of endogenous proteins involves peptide transport to the endo-lysosomal compartment. J Cell Sci 2004; 117: 4219–4230.
Jonjić S, Babić M, Polić B, Krmpotić A . Immune evasion of natural killer cells by viruses. Curr Opin Immunol 2008; 20: 30–38.
Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod'homme V, Aicheler R et al. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol 2008; 41: 206–212.
Rao P, Pham HT, Kulkarni A, Yang Y, Liu X, Knipe DM et al. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol 2011; 85: 8093–8104.
Brigl M, Brenner MB . CD1: antigen presentation and T cell function. Annu Rev Immunol 2004; 22: 817–890.
del Val M, Hengel H, Häcker H, Hartlaub U, Ruppert T, Lučin P et al. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J Exp Med 1992; 176: 729–738.
Ziegler H, Thäle R, Lučin P, Muranyi W, Flohr T, Hengel H et al. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 1997; 6: 57–66.
Hengel H, Reusch U, Gutermann A, Ziegler H, Jonjić S, Lučin P et al. Cytomegaloviral control of MHC class I function in the mouse. Immunol Rev 1999; 168: 167–176.
Kleijnen MF, Huppa JB, Lučin P, Mukherjee S, Farrell H, Campbell AE et al. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J 1997; 16: 685–694.
Reusch U, Muranyi W, Lučin P, Burgert HG, Hengel H, Koszinowski UH . A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J 1999; 18: 1081–1091.
Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH . Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med 2002; 16: 805–816.
Ilić Tomaš M, Kučić N, Mahmutefendić H, Blagojević G, Lučin P . Murine cytomegalovirus perturbs endosomal trafficking of MHC class I molecules in the early phase of infection. J Virol 2010; 84: 11101–11112.
Mahmutefendić H, Blagojević G, Kučić N, Lučin P . Constitutive internalization of murine MHC class I molecules. J Cell Physiol 2007; 210: 445–455.
Kučić N, Ilić Tomaš M, Mahmutefendić H, Blagojević G, Lučin P . Early endosomal retention of murine cytomegalovirus m06 protein. Croat Chem Acta 2012; 85: 213–221.
Lenac T, Budt M, Arapovic J, Hasan M, Zimmermann A, Simic H et al. The herpesviral Fc receptor fcr-1down-regulates the NKG2D ligands MULT-1 and H60. J Exp Med 2006; 203: 1843–1850.
Jefferies WA, Burgert GH . E3/19K from adenovirus 2 is an immunosubversive protein that binds to a structural motif regulating the intracellular transport of major histocompatibility complex class I proteins. J Exp Med 1990; 172: 1653–1664.
Sanchez V, Greis KD, Sztul E, Britt WJ . Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 2000; 74: 975–986.
Homman-Loudiyi M, Hultenby K, Britt W, Soderberg-Naucler C . Envelopment of human cytomegalovirus occurs by budding into Golgi derived vacuole compartments positive for gB, Rab 3, trans-Golgi network 46, and mannosidase II. J Virol 2003; 77: 3191–3203.
Fraile-Ramos A, Pelchen-Matthews A, Kledal TN, Browne H, Schwartz TW, Marsh M . Localization of HCMV UL33 and US27 in endocytic compartments and viral membranes. Traffic 2002; 3: 218–232.
Seo JY, Britt WJ . Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J Virol 2006; 80: 5611–5626.
Das S, Vasanji A, Pellett PE . Three dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 2007; 81: 11861–11869.
Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T et al. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol 2011; 85: 3821–3832.
Tandon R, Mocarski ES . Viral and host control of cytomegalovirus maturation. Trends Microbiol 2012; 20: 392–401.
Cepeda V, Esteban M, Fraile-Ramos A . Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol 2010; 12: 386–404.
Das S, Pellett PE . Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 2011; 85: 5864–5879.
Hertel L, Mocarski ES . Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle expression and induction of pseudomitosis independent of US28 function. J Virol 2004; 78: 11988–12011.
Tooze J, Hollinshead M, Reis B, Radsak K, Kern H . Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur J Cell Biol 1993; 60: 163–178.
Fraile-Ramos A, Cepeda V, Elstak E, van der Sluijs P . Rab27a is required for human cytomegalovirus assembly. PLoS One 2010; 5: e15318.
Krzyzaniak MA, Mach M, Britt WJ . HCMV-encoded glycoprotein M (UL100) interacts with Rab11 effector protein FIP4. Traffic 2009; 10: 1439–1457.
Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M et al. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 2014; 15: 363–373.
Hengel H, Koopmann JO, Flohr T, Muranyi W, Goulmy E, Hammerling GJ et al. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 1997; 6: 623–632.
Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL . The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996; 84: 769–779.
Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR et al. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996; 384: 432–438.
Ahn K, Angulo A, Ghazal P, Peterson PA, Yang Y, Fruh K . Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc Natl Acad Sci USA 1996; 93: 10990–10995.
Jones TR, Wiertz EJ, Sun L, Fish KN, Nelson JA, Ploegh HL . Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc Natl Acad Sci USA 1996; 93: 11327–11333.
Barnes PD, Grundy JE . Down-regulation of the class I HLA heterodimer and β2-microglobulin on the surface of cells infected with cytomegalovirus. J Gen Virol 1992; 73: 2395–2403.
Fairley JA, Baillie J, Bain M, Sinclair JH . Human cytomegalovirus infection inhibits epidermal growth factor (EGF) signalling by targeting EGF receptors. J Gen Virol 2002; 83: 2803–2810.
Hassink GC, Duijvestijn-van Dam JG, Koppers-Lalic D, van Gaans-van den Brink J, van Leeuwen D, Vink C et al. Rat cytomegalovirus induces a temporal downregulation of major histocompatibility complex class I cell surface expression. Viral Immunol 2005; 18: 607–615.
Baca Jones CC, Kreklywich CN, Messaoudi I, Vomaske J, McCartney E, Orloff SL et al. Rat cytomegalovirus infection depletes MHC II in bone marrow derived dendritic cells. Virology 2009; 388: 78–90.
Emery VC, Cope AV, Bowen EF, Gor D, Griffiths PD . The dynamics of human cytomegalovirus replication in vivo. J Exp Med 1999; 190: 177–182.
Riegler S, Hebart H, Einsele H, Brossart P, Jahn G, Sinzger C . Monocyte-derived dendritic cells are permissive to the complete replicative cycle of human cytomegalovirus. J Gen Virol 2000; 81: 393–399.
Raftery MJ, Schwab M, Eibert SM, Samstag Y, Walczak H, Schonrich G . Targeting the function of mature dendritic cells by human cytomegalovirus: a multilayered viral defense strategy. Immunity 2001; 15: 997–1009.
Odeberg J, Soderberg-Naucler C . Reduced expression of HLA class II molecules and Iinterleukin-10- and transforming growth factor beta1-independent suppression of T-cell proliferation in human cytomegalovirus-infected macrophage cultures. J Virol 2001; 75: 5174–5181.
Spencer JV, Lockridge KM, Barry PA, Lin G, Tsang M, Penfold ME et al. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol 2002; 76: 1285–1292.
Lee AW, Hertel L, Louie RK, Burster T, Lacaille V, Pashine A et al. Human cytomegalovirus alters localization of MHC class II and dendrite morphology in mature Langerhans cells. J Immunol 2006; 177: 3960–3971.
Cebulla CM, Miller DM, Zhang Y, Rahill BM, Zimmerman P, Robinson JM et al. Human cytomegalovirus disrupts constitutive MHC class II expression. J Immunol 2002; 169: 167–176.
Loewendorf AI, Steinbrueck L, Peter C, Busche A, Benedict CA, Kay-Jackson PC . The mouse cytomegalovirus glycoprotein m155 inhibits CD40 expression and restricts CD4 T cell responses. J Virol 2011; 85: 5208–5212.
Mintern JD, Klemm EJ, Wagner M, Paquet ME, Napier MD, Kim YM et al. Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus Fc receptor-1. J Immunol 2006; 177: 8422–8431.
Moutaftsi M, Mehl AM, Borysiewicz LK, Tabi Human Z . cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood 2002, 99: 2913–2921.
Hertel L, Lacaille VG, Strobl H, Mellins ED, Mocarski ES . Susceptibility of immature and mature Langerhans cell-type dendritic cells to infection and immunomodulation by human cytomegalovirus. J Virol 2003; 77: 7563–7574.
Beck K, Meyer-König U, Weidmann M, Nern C, Hufert FT . Human cytomegalovirus impairs dendritic cell function: a novel mechanism of human cytomegalovirus immune escape. Eur J Immunol 2003; 33: 1528–1538.
Reeves M, Sinclair J . Regulation of human cytomegalovirus transcription in latency: beyond the mayor immediate-early promoter. Viruses 2013; 5: 1395–1413.
Slobedman B, Mocarski ES, Arvin AM, Mellins ED, Abendroth A . Latent cytomegalovirus down-regulates major histocompatibility complex class II expression on myeloid progenitors. Blood 2002; 100: 2867–2873.
Bacon L, Eagle RA, Meyer M, Easom N, Young NT, Trowsdale J . Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J Immunol 2004; 178: 21078–21084.
Nachmani D, Lankry D, Wolf DG, Mandelboim O . The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol 2010; 11: 806–813.
Müller S, Zocher G, Steinle A, Stehle T . Structure of the HCMV UL16–MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PloS Pathog 2010; 6: 1000723.
Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR . NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 2009; 83: 12345–12354.
Fielding CA, Aicheler R, Stanton RJ, Wang EC, Han S, Seirafian S . Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog 2014; 10: e1004058.
Aguera-Gonzalez S, Boutet P, Reyburn HT, Vales-Gomez M . Brief residence at the plasma membrane of the MHC class I-related chain B is due to clathrin-mediated cholesterol-dependent endocytosis and shedding. J Immunol 2009; 182: 4800–4808.
Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995; 375: 411–415.
Früh K, Ahn K, Djaballah H, Sempé P, van Endert PM, Tampé R et al. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995; 375: 415–418.
Levitskaya J, Coram M, Levitsky V, Imreh S, Steigerwald-Mullen PM, Klein G et al. Inhibition of antigen processing by the internal repeat region of the Epstein–Barr virus nuclear antigen-1. Nature 1995; 375: 685–688.
Kwun HJ, da Silva SR, Shah IM, Blake N, Moore PS, Chang Y . Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein–Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J Virol 2007; 81: 8225–8235.
Bennett NJ, May JS, Stevenson PG . Gamma-herpesvirus latency requires T cell evasion during episome maintenance. PLoS Biol 2005; 3: e120.
Abendroth A, Lin I, Slobedman B, Ploegh H, Arvin AM . Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J Virol 2001; 75: 4878–4888.
Hudson AW, Howley PM, Ploegh HL . A human herpesvirus 7 glycoprotein, U21, diverts major histocompatibility complex class I molecules to lysosomes. J Virol 2001; 75: 12347–12358.
May NA, Glosson NL, Hudson AW . HHV-7 U21 downregulates classical and non-classical class I MHC molecules from the cell surface. J Virol 2010; 84: 3738–3751.
Schneider CL, Hudson AW . The Human Herpesvirus-7 (HHV-7) U21 immunoevasin subverts NK-mediated cytotoxicity through modulation of MICA and MICB. PLoS Pathog 2011; 7: e1002362.
Sullivan BM, Coscoy L . Downregulation of the T cell receptor complex and impairment of T-cell activation by human herpesvirus 6 U24 protein. J Virol 2008; 82: 602–608.
Yuan W, Dasgupta A, Cresswell P . Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol 2006; 7: 835–842.
Raftery MJ, Winau F, Kaufmann SH, Schaible UE, Schönrich G . CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol 2006; 177: 6207–6214.
Imai T, Koyanagi N, Ogawa R, Shindo K, Suenaga T, Sato A et al. Us3 kinase encoded by herpes simplex virus 1 mediates downregulation of cell surface major histocompatibility complex class I and evasion of CD8+ T cells. PLoS ONE 2013; 8: e72050.
Rappocciolo G, Birch J, Ellis SA . Down-regulation of MHC class I expression by equine herpesvirus-1. J Gen Virol 2003; 84: 293–300.
Said A, Azab W, Damiani A, Osterrieder N . Equine herpesvirus type 4 UL56 and UL49.5 proteins downregulate cell surface major histocompatibility complex class I expression independently of each other. J Virol 2012; 86: 8059–8071.
Koppers-Lalic D, Rijsewijk FA, Verschuren SB, van Gaans-Van den Brink JA, Neisig A, Ressing ME et al. The UL41-encoded virion host shutoff (vhs) protein and vhs-independent mechanisms are responsible for down-regulation of MHC class I molecules by bovine herpesvirus 1. J Gen Virol 2001; 82: 2071–2081.
Coscoy L, Ganem D . Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA 2000; 97: 8051–8056.
Ishido S, Wang C, Lee BS, Cohen GB, Jung JU . Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J Virol 2000; 74: 5300–5309.
Duncan LM, Piper S, Dodd RB, Saville MK, Sanderson CM, Luzio JP et al. Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J 2006; 25: 1635–1645.
Hewitt EW, Duncan L, Mufti D, Baker J, Stevenson PG, Lehner PJ . Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J 2002; 21: 2418–2429.
Coscoy L, Ganem D . A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J Clin Invest 2001; 107: 1599–1606.
Sanchez DJ, Gumperz JE, Ganem D . Regulation of CD1d expression and function by a herpesvirus infection. J Clin Invest 2005; 115: 1369–1378.
Thomas M, Wills M, Lehner PJ . Natural killer cell evasion by an E3 ubiquitin ligase from Kaposi's sarcoma-associated herpesvirus. Biochem Soc Trans 2008; 36: 459–463.
Mansouri M, Douglas J, Rose PP, Gouveia K, Thomas G, Means RE et al. Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 2006; 108: 1932–1940.
Bartee E, McCormack A, Fruh K . Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog 2006; 2: e107.
Li Q, Means R, Lang S, Jung JU . Downregulation of gamma interferon receptor 1 by Kaposi's sarcoma-associated herpesvirus K3 and K5. J Virol 2007; 81: 2117–2127.
Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME et al. The Epstein–Barr virus G-protein coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog 2009; 5: e1000255.
Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse HJ et al. The Epstein–Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J Virol 2011; 85: 1604–1614.
Morrow G, Slobedman B, Cunningham AL, Abendroth A . Varicella zoster virus productively infects mature dendritic cells and alters their immune function. J Virol 2003; 77: 4950–4959.
Acknowledgements
This work was supported by the Ministry of Science, Education and Sport of the Republic of Croatia (grants 062006, 062-0620238-0223 and 0062030), the European FP6 EMBIC project (No. 512040, LSHM-CT-2004-512040) and by the University of Rijeka (grants 13.06.1.1.4, 13.06.2.1.55 and 13.06.2.1.56).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no financial or commercial conflicts of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0
About this article

Cite this article
Lučin, P., Mahmutefendić, H., Blagojević Zagorac, G. et al. Cytomegalovirus immune evasion by perturbation of endosomal trafficking. Cell Mol Immunol 12, 154–169 (2015). https://doi.org/10.1038/cmi.2014.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2014.85
Keywords
This article is cited by
-
Function of the cargo sorting dileucine motif in a cytomegalovirus immune evasion protein
Medical Microbiology and Immunology (2019)
-
Distinct expression profile of HCMV encoded miRNAs in plasma from oral lichen planus patients
Journal of Translational Medicine (2017)
-
Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome
Virology Journal (2017)
-
CMV immunology
Cellular & Molecular Immunology (2015)
-
An endocytic YXXΦ (YRRF) cargo sorting motif in the cytoplasmic tail of murine cytomegalovirus AP2 ‘adapter adapter’ protein m04/gp34 antagonizes virus evasion of natural killer cells
Medical Microbiology and Immunology (2015)
