
Abstract
Major mechanisms for the recognition of pathogens by immune cells have evolved to employ classical and non-classical major histocompatibility complex class I (MHC I) molecules. Classical MHC I molecules present antigenic peptide ligands on infected cells to CD8+ T cells, whereas a key function for non-classical MHC I molecules is to mediate inhibitory or activating stimuli in natural killer (NK) cells. The structural diversity of MHC I puts immense pressure on persisting viruses, including cytomegaloviruses. The very large coding capacity of the human cytomegalovirus allows it to express a whole arsenal of immunoevasive factors assigned to individual MHC class I targets. This review summarizes achievements from more than two decades of intense research on how human cytomegalovirus manipulates MHC I molecules and escapes elimination by the immune system.
Similar content being viewed by others


Major histocompatibility complex class I molecules
Major histocompatibility complex (MHC) class I molecules represent a basic molecular framework that mediates the activation and function of cytotoxic effector cells of the adaptive and innate branches of the immune system, such as CD8+ T cells and natural killer (NK) cells. MHC I molecules are cell surface resident type I transmembrane glycoproteins that possess a common molecular architecture with an α1 and an α2 domain (Figure 1). Most also contain an α3 domain and are able to dimerize with soluble β-2-microglobulin (β2m) (Figure 1). The need to load a peptide of 8 to 10 amino acids (aa) in length between the α1 and α2 domains in order to stabilize the protein conformation is a hallmark of polymorphic (‘classical') MHC I molecules (Table 1). The eminently developed polymorphic nature of the classical MHC I molecules is reflected by a very high number of HLA-A, -B and -C alleles. Their strict peptide dependency has allowed the CD8+ T-cell arm of the adaptive immune system to develop, providing long-lasting antigen-specific immunological memory to the host.

Domain structure of MHC I molecules highlighted on the background of HLA-B*07:02 loaded with a pp65 peptide.164 The α1 and α2 domains are shown in blue and the conserved α3 and β2m in green and yellow, respectively. The pp65 peptide is depicted in a ball-and-stick model with the N-terminus pointing towards the viewer. MHC, major histocompatibility complex.
CD8+ T cells express T-cell receptors (TCRs) that are activated in a highly sensitive manner through low affinity binding to the peptide bound to MHC class I molecules. In most cell types, MHC I molecules gain their peptide ligands from endogenous polypeptide degradation products, thereby sampling the cellular protein content for CD8+ T cells. The T cell is activated upon the recognition of a foreign peptide on a MHC I complex, leading to elimination of the target cell. Many viruses have evolved mechanisms to escape this recognition process by downregulating the peptide presenting MHC class I molecules from the cell surface. Because of this, the recognition algorithm needs to be complemented by an independent surveillance system responding to the absence of MHC I molecules (‘missing self').1 To this end, the human immune system has produced particular killer cell immunoglobulin-like receptors (KIRs) and leukocyte Ig-like receptors (LIRs) expressed by NK cells (and by other cells types that are discussed later). Both receptors bind to MHC I molecules and inhibit NK cell activation. Consequently, if expression of MHC I is impaired, for example by viruses, the inhibitory signal is reduced, facilitating the activation of signaling for ‘missing self' and resulting in cytotoxic destruction of the target cell.
From this description, it is clear that classical peptide presentation by MHC I molecules serves a complex homeostatic mechanism as a ligand for TCRs expressed by CD8+ T cells, and also by KIRs and LIRs expressed by NK cells. Both tasks are grossly distributed between three MHC I genes encoded in the MHC locus on human chromosome 6. Whereas all HLA-C alleles possess the ability to bind to KIRs, only one third of HLA-A and HLA-B alleles are able to engage them (reviewed in Ref. 2). In view of this fact, HLA-A and -B complexes are regarded as the more potent antigen presenting MHC I molecules. The HLA-A and -B genes are also more variable than the HLA-C gene, with HLA-B being the most polymorphic gene known in humans. Nevertheless, a number of CD8+ T-cell epitopes presented by HLA-C molecules have been identified,3 exemplifying the high plasticity of MHC I molecules and their physiological ‘dual use' (i.e., being both antigen presenters to CD8+ T cells and ligands for NK cell receptors).
Depending on the allele specificity of the MHC I binding receptors, contact sites with different degrees of conservation are engaged on MHC I. The CD8+ co-receptor binding to MHC I is allele-independent, demonstrated by the recognition of conserved regions predominantly in the α3 domain of MHC I but also in α2 and β2m.4 This is also the case for LIR1 (also called ILT-2 or CD85j), which contacts conserved α3 and β2m residues,5 thereby rendering this interaction possibly even less dependent on the MHC I allele. Indeed, LIR-1 has been verified to interact with HLA-A, -B, -C, -G and -F molecules.6,7,8 KIRs contact the C-terminal α1 domain and adjacent regions on the α2 domain. In some instances, the loaded peptide will confer specificity to the KIR–MHC I interaction.9 These less conserved determinants of MHC I confer a more stringent allele specificity for KIRs than for LIR receptors. Accordingly, the contact sites of the TCR on both the α1 and α2 domains of MHC I and especially on the loaded peptide render the TCR interaction with MHC I the most selective.10
In addition to receptors recognizing classical MHC I, NK cells express a large arsenal of prominent inhibitory and activating receptors (e.g., NKG2A, NKG2C, NKG2D; Table 1), that are capable of interacting with a broad spectrum of different ligands. Many of these are non-classical MHC I molecules, such as MHC class I chain-related (MIC) A and MICB and the UL16-binding proteins (ULBP), all of which are induced in target cells by stress signals.11 These non-classical MHC I molecules are common targets for viral immunoevasins that both downregulate activating ligands and induce inhibitory signals in NK cells. This review summarizes our current knowledge of the intertwined and sophisticated mechanisms by which human cytomegalovirus (HCMV) targets classical and non-classical MHC I molecules to counteract cytotoxic effector cells and, in some cases, to restore immune recognition.
HCMV
HCMV constitutes a prototypical β-herpesvirus within the human pathogenic Herpesviridae family. Herpes viruses are found in all human populations, with high immunoglobulin G (IgG) seroprevalence rates of 50%–98%.12 Despite the expression of a very large antigenic proteome of about 750 translational products13 during the sequentially ordered immediate-early (IE), early (E) and late (L) phases of gene expression, HCMV avoids sterile immunity and persists lifelong in the human host. The virus exists in a latent state, with periodic phases of reactivation, lytic replication and virus shedding. The co-evolution and co-speciation of HCMV with the human host over millions of years has equipped the virus with greatly adapted mechanisms for the evasion and exploitation of human immune functions. Nonetheless, HCMV replication is well controlled by the healthy immune system, causing overt disease only after failure of essential components of antiviral control, such as CD8+ T cells or NK cells. As a consequence, the HCMV disease burden is closely associated with multiple conditions of immunocompromisation and immunological immaturity, with the latter explaining severe disease manifestations observed in congenitally infected infants.12,14 An astounding hallmark of HCMV infections is the prodigious expansion of CD8+ T cells specific for only a few HCMV epitopes. This subpopulation can comprise more than 20% of the CD8+ T-cell memory compartment.15 Likewise, cytomegalovirus exerts a strong imprinting effect on subtypes of NK cells bearing CD94/NKG2C and/or specific KIRs and promotes the expansion of these cells.16,17
Assembly of classical MHC class I molecules
Classical MHC I molecules consist of the heavy chain comprising the α1-3 ectodomains, a transmembrane segment (TMS) and a cytosolic tail. Assisted by consecutive interactions with the chaperones calnexin and calreticulin, the heavy chain is folded and dimerizes with β2m in the lumen of the endoplasmic reticulum (ER), thereby forming a peptide receptive complex that can be recruited to the peptide loading complex (PLC). The PLC is a multimolecular complex that assembles around the dimeric peptide transporter TAP (transporter associated with antigen processing), consisting of the subunits TAP1 and TAP2. In addition to the formation of the peptide translocation pore, both TAP subunits possess N-terminal segments in their transmembrane domains that contain binding sites for tapasin,18,19,20 important for the stabilization of the TAP dimer and induction of peptide transport.21,22 Furthermore, by binding to MHC I molecules, tapasin places MHC I into close vicinity to TAP1/2. After TAP transport, N-terminally extended MHC I precursor peptides are trimmed by ER aminopeptidases to obtain a length fitting the MHC I peptide binding groove.23 Subsequently, the tapasin-MHC I interaction facilitates efficient loading of peptide ligands, thereby inducing a stable conformation and the ability of MHC I to exit from the ER and be transported to the cell surface. Further chaperones participating in the peptide-loading procedure in the PLC are the lectin-like chaperone calreticulin and the oxidoreductase ERp57. It has been suggested that the unique stable dimer that is formed between ERp57 (Cys57) and tapasin (Cys95) is important for quality control of MHC I peptide loading.24 The first 87 N-terminal amino acids of tapasin are sufficient to facilitate effective peptide loading on recombinant MHC I molecules,25 but the entire mechanistic role of tapasin and ERp57 in the process of MHC I quality control remains to be uncovered.
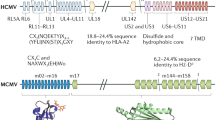
HCMV interference with classical MHC I molecules
Early studies in the 1990s indicated that HCMV targets MHC I molecules and their functions along the antigen presentation pathway in infected cells at numerous points during the maturation process.26,27,28,29,30,31,32 The identification of the responsible viral molecules (collectively called ‘immunoevasins'33) provided unique tools to elucidate molecular mechanisms, structures and important compartmental checkpoints of the MHC I antigen presentation pathway. Four inhibitors of the HCMV US6 gene family are sufficient for strong downregulation of MHC I molecules from the cell surface upon gene transfection and were extensively studied upon their discovery; these inhibitors include US2, US3, US6 and US11 (Table 1). Whereas US2 and US11 are expressed as early proteins that target MHC I molecules for proteasomal degradation, US3 represents an immediate early protein that retains MHC I in the ER (Figure 2).34,35,36,37 US6 is synthesized during early and late infection and inhibits peptide loading by blocking the peptide transporter TAP (Figure 2).32,38,39,40 All US6 gene family members are type I transmembrane proteins with a non-conserved N-terminal stretch in front of an Ig-like domain formed by conserved cysteines,41 a TMS and a cytosolic tail. Thus, the molecules share several homologies, with basic structural arrangements most likely preserved and a conserved membrane topology.41 However, the not very well-conserved primary sequences reflect considerable functional diversity and different protein interactions.

Inhibition of classical MHC I molecules during IE, E and E/L phases of HCMV replication. Under IE conditions of HCMV infection only US3 (blue) is expressed. By forming oligomers, US3 binds to MHC I, leading to MHC I retention in the ER. Additionally, US3 binds to TAP1 and tapasin and blocks interaction of PLC components, thereby conceivably inhibiting proper peptide loading of MHC I molecules. In the E phase, US2 (red) and US11 (yellow) are expressed and direct MHC I molecules to ERAD pathways for proteosomal degradation in the cytosol. US2 and US11 utilize, SPP/TRC8 and Derlin-1/TMEM129 ERAD pathways, respectively. Circumstantial evidence suggests the action of an additional not yet identified factor (light blue) interfering with MHC I recruitment to the PLC. In conjunction with US6 (green), the factors continue to be expressed during the late (L) phase of infection. US6 forms oligomers and blocks TAP-dependent peptide translocation into the ER. ER, endoplasmic reticulum; ERAD, ER-associated degradation; HCMV, human cytomegalovirus; MHC, major histocompatibility complex; PLC, peptide loading complex.
US2
Of the four mentioned inhibitors, only the structure of the US2 protein in complex with HLA-A*02:01 has been resolved41 to date. A soluble US2 mutant corresponding to aa 15–140 of its luminal domain was sufficient to form a stable complex with the MHC I molecule.42 In this crystal structure, the N-terminal residues 15–42 of US2 were not resolved, indicating that these residues are flexible or unstructured. This part of US2 has also been shown to be dispensable for US2 function.43 Interestingly, the same soluble mutant of US2 did not form a complex with the HLA-B alleles B*27 and B*07,42 even though HLA-B*27 is targeted for degradation by wild type US2.44 This implies that US2 is able to contact MHC I molecules in different manners. Indeed, whereas the HLA-A*02:01 residues contacted by US2 in the regions 105–107 and 264–267 are more conserved between MHC I alleles, a third region comprising aa 176–183 is polymorphic, suggesting that US2 must be able to overcome differences in this region of MHC I by displaying flexible binding modes dependent on the target MHC I allele.41 These sequence differences possibly render the interaction between US2 and MHC I more stable in the case of HLA-A2 compared with HLA-B27; however, this does not exclude that different modes of interaction will meet the same final fate of MHC I degradation.
The residues contacted by US2 are located in a region between the C-terminus of the α2 helix and the α3 domain and are therefore distant from PLC contact sites on MHC I.45 Hence, binding site competition is avoided and allows US2 to attack MHC I molecules that are already associated with the PLC.
Several studies found that HLA-B*07 escapes US2-mediated degradation.44,46,47 Based on HLA-B*07 resistance towards US2, MHC I allele sequence analysis predicted that US2 resistance should also be conveyed by HLA-B*08, B*40, B*41, B*42 and B*48.44 Interestingly, in cells infected with an HCMV deletion mutant expressing only US2 and not US3, US6 or US11, HLA-B*07 was efficiently downregulated, but HLA-B*08 was not,48 which is in clear contrast to the abovementioned studies. This discrepancy could indicate that US2 is functionally different in the context of HCMV infection when compared with selective US2 expression systems.
Whereas the ectodomain of US2 is responsible for the initial recognition of target MHC I molecules, its TMS and cytosolic tail are necessary to complete subsequent steps of the interaction, such as forwarding MHC I for proteasomal degradation.49 Analysis of a lysine-free HLA-A*02 allele revealed that the ubiquitination of the target MHC I molecule is a prerequisite for US2-mediated dislocation and degradation in the cytosol.50 The E3 ligase utilized by US2 was identified to be TRC8 (translocation in renal carcinoma, chromosome 8 gene), a multimembrane-spanning, ER-resident RING-type E3-ligase. In cells lacking TRC8, US2 is not able to degrade MHC I.51 It was suggested that US2 interacts with the E3 ligase via its TMS. Prior to the identification of TRC8, it was shown that the signal peptide peptidase (SPP) is a necessary component of the US2-dependent MHC I dislocation complex, even though the role for SPP has not yet been clearly defined. Normally, SPP is responsible for cleaving off the signal peptide of proteins that are targeted to the ER lumen via the Sec61 translocon complex. It was demonstrated that SPP and TRC8 interact not only in the presence but also in the absence of US2, suggesting that these molecules could work in concert to dispose signal peptides and that US2 exploits this mechanism to prohibit MHC I antigen presentation.51 A further interesting observation is that the signal peptide of US2 is not cleaved off after translocation into the ER.43 Although the cytosolic tail of US2 is sufficient for interaction with SPP,52 it could be envisaged that the signal peptide of US2 plays an additional role in recruiting SPP to the site of interaction with MHC I. US2 is indeed itself a substrate for proteasomal degradation in the cytosol.43
Additionally, it was observed that lack of protein disulfide isomerase reduced the degradation levels of MHC I and conversely induced the interaction with US2, suggesting that protein disulfide isomerase might be important for the disruption of the interaction between MHC I and US2 prior to MHC I dislocation.53
The target specificity for US2 expands beyond classical MHC I molecules: US2 has been shown to degrade HLA-G, HFE, the MHC class II chains DR-α and DM-α and possibly also CD1d in a similar manner to classical MHC I molecules.44,54,55,56,57 The question remains whether US2 attacks these diverse targets directly or if it takes advantage of adaptor proteins linking their substrates to US2-controlled degradation pathways.
US11
In a landmark study by the Ploegh laboratory, US11 was the first HCMV protein described to exploit an ER-associated degradation (ERAD) pathway in order to block MHC I antigen presentation (Figure 2).58 What at a first glance seems to be a redundant function with the concomitantly expressed US2 inhibitor during the extended E phase of the HCMV replication cycle turned out to be a complementing and elaborate immunoevasive strategy.
As observed with US2, US11 is able to recognize both free and β2m-associated MHC I heavy chains.46 However, US11-mediated MHC I degradation utilizes a different route than US2. Instead of taking advantage of SPP and TRC8, US11 directs MHC I to ERAD degradation through a Derlin-1-dependent pathway.59,60 Deploying its ectodomain, US11 interacts with MHC I molecules61,62 and recruits the intramembrane pseudoprotease Derlin-1 via a single amino acid in the TMS (Q192).59,60,62 The transfer of MHC I to Derlin-1 is dependent on the length of the MHC I cytosolic tail.63 Specific hydrophobic residues at the terminus of the cytosolic tail lead to efficient recruitment of the dislocation complex,64 which consists of VIMP,60 the AAA ATPase p97,65 and SEL1L.66 Upon ubiquitination of the MHC I molecule, it becomes rapidly dislocated and degraded. In two recent independent studies, the dislocation complex was resolved in even greater detail; TMEM129, a previously uncharacterized protein, was shown to be the E3 ligase utilized by US11.67,68 In cells deficient in TMEM129 expression, the US11-dependent degradation of MHC I was lost. Both studies demonstrated that an interaction between TMEM129 and Derlin-1 also occurs in the absence of US11 and in conjunction with the increased unfolded protein response occurring in TMEM129-deficient cells, this implies that TMEM129 is a true and essential component of ERAD. Furthermore, it was shown that both the previously described E2 ligase UBE2K (E2-25K)67,69 and a newly identified UBE2J2 are required for US11-mediated degradation of MHC I.67,68 Therefore, in a typical E2 enzyme reaction pattern, UBE2J2 possibly confers the initiation of MHC I monoubiquitination, whereas UBE2K promotes the elongation of the ubiquitin chain.67
Also US11 exhibits MHC I allele-specific effects. Whereas it is well documented that HLA-A2 is rapidly degraded in the presence of US1170 and HLA-C is resistant to US11,71 the effect of US11 on HLA-B alleles is not yet clear. In addition to the cytosolic tail of MHC I,63,70 degradation efficiency depends on luminal interactions between US11 and MHC I.63 Data based on the analysis of swap mutants of the α1 and α2 domains of HLA-A2 and HLA-G suggested that US11 recognizes specific determinants of these domains.63 On the contrary, antigen presentation by the US11-sensitive allele HLA-A*02:01 became resistant to US11 when a chimera of HLA-A*02:01 and HLA-C*07:02 consisting of the α1 and α2 domains from HLA-*02:01 was applied. In contrast, a chimera with α1 and α2 domains corresponding to HLA-C*07:02 but with a C-terminal HLA-A*02:01 sequence was sensitive towards US11.72 However, neither essential residues on MHC I nor on US11 have been defined that are important for the US11-MHC I interaction. Most likely, US11 recognizes multiple structural determinants in a hierarchical or ‘proofreading' manner.
It should be noted that the TMS of US11 is highly potent in directing interaction partners for degradation. Sole expression of the US11 TMS in the context of HLA-A2 and the vesicular stomatitis virus glycoprotein forced interactions with Derlin-1 and rendered them highly unstable and sensitive to proteasomal degradation.73 Clearly, US11 must in itself possess mechanisms to resist Derlin-1-dependent degradation. Indeed, a very recent study showed that swapping the HLA-A2 cytosolic tail for the US11 molecule renders US11 highly sensitive to degradation.68
US3
The impact of US3 on MHC I is fully different from US2 and US11. Expressed as an immediate early protein for only a few hours after infection,27 US3 retains MHC I molecules for an extensive period of time in the ER (Figure 2). However, the final fate of these molecules has not been determined. Several studies suggest that even if delayed, a large portion of the MHC I molecules will finally reach the cell surface.36,74,75 In contrast to US11, US3 does not trigger NK cell activation by MHC I downregulation.75 Whether the surface expressed MHC I molecules have ‘slipped' out of US3 control or whether this is a deliberate intention of the US3-mediated inhibition strategy is not clear. Possibly only certain MHC I alleles are retained, while others escape the US3 effect.36 US3 was shown to bind to tapasin and TAP and to reduce the level of interaction between the PLC components.76 This interaction could result in reduced MHC I quality control. Whereas tapasin-independent MHC I alleles were not affected by this US3 function, the maturation and surface expression of tapasin-dependent MHC I were hindered.76 It could be envisaged that MHC I molecules reaching the cell surface do not possess optimal antigen presentation potential, but rather present HCMV-controlled decoy molecules. Moreover, in HCMV infected cells US3 possibly works in concert with other immunoevasins, which could change its net effect on MHC I provided that the immunoevasins are co-expressed at a given timepoint of the protracted HCMV replication cycle. A synergistic effect on MHC I downregulation has already been demonstrated for US3 and US11.77
Despite a short half-life of approximately one hour, US3 gains Endo H resistance and is transported to lysosomes, where it is degraded.36,74 Therefore, newly synthesized US3 molecules are required to encounter MHC I in the ER.74 It has been suggested that US3 forms flexible oligomers to which newly synthesized US3 polypeptides are recruited to prevent MHC I from leaving the ER.78 Three residues (Ser 58, Glu 63 and Lys 64) in the ectodomain of US3 were found to be important for US3 retention.79 Single mutation of one of the three residues was sufficient to interrupt ER localization and MHC I retention, but not interaction with MHC I.79 Both the ectodomain and the TMS of US3 are essential in order to preserve the interaction with MHC I.80
Notably, two forms of US3 have been observed. The longer variant is approximately 22 kDa and exerts the inhibitory effect on MHC I. The shorter form of US3 is approximately 18 kDa and lacks a TMS.36 It was shown that the expression of the shorter form in HeLa cells releases MHC I from the inhibitory effect by the longer US3 form, suggesting that the shorter US3 variant is able to regulate the magnitude of MHC I control.81
US6
US6, expressed with early/late kinetics during the HCMV replication cycle, does not target MHC I directly, but blocks peptide transport by the peptide transporter TAP.38,39,40 Controlling the import of a large majority of peptides into the ER, TAP inhibition leads to efficient downregulation of all peptide dependent classical and non-classical MHC I molecules.82 In contrast, HLA-A*02:01 is exceptionally resistant to TAP inhibition, and peptides produced in a TAP-independent manner can stabilize HLA-A*02:01 and confer cell surface expression of this allele.83 UL40 counteracts US6 by providing its signal peptide for HLA-E and UL18 stabilization84,85 (discussed later).
Like other US6 gene family members, US6 is a glycosylated type I transmembrane protein and blocks TAP by its ectodomain (i.e., the TMS and cytosolic tail of US6 are dispensable for its function).38 Anchoring of US6 to the membrane renders the protein substantially more efficient (unpublished observation). TAP function is dependent on ATP in order to complete a cycle of peptide translocation.86 By binding to TAP, US6 inhibits crosstalk between the transmembrane domains and the cytosolic nucleotide domains of both TAP subunits; binding of ATP to TAP is blocked and peptide translocation cannot proceed.87 By the insertion of a point mutation in the ectodomain of US6 (Cys108Tyr), US6 lost the ability to block mouse and rat TAP dimers.88 This instrumental mutation was utilized to define US6 contact sites on TAP1 and TAP2, thereby experimentally revealing the 10 TMS topology of the TAP1 subunit.89 By forming oligomers, US6 interacts with at least four independent sites formed by the TAP dimer, the significance of which differs with regard to the inactivation of the transporter.89 In accordance with this, US6 was shown to possess a core domain indispensable for TAP inhibition but not for binding (aa 89–108), and also a site proximal to the ER membrane (aa 116–125) that conferred stability to the TAP interaction and enhanced the inhibitory effect.90
Other HCMV regulated functions that might affect classical MHC I molecules in HCMV-infected cells
It is conceivable that in addition to US2, US3, US6 and US11, additional proteins impact MHC I antigen presentation in HCMV-infected cells. In a closely related non-human primate cytomegalovirus (rhesus cytomegalovirus), in addition to the US2 to US11 genes another potent MHC I inhibitor was identified (Rh178) that is not conserved between HCMV and rhesus cytomegalovirus.91
We observed that tapasin biosynthesis is impaired independently of the HCMV US2–US6 genes during the course of HCMV replication, and that MHC I molecules are inefficiently recruited to the PLC,92 pointing at interference with the process of MHC I peptide loading. Because these phenotypes cannot be explained by the established inhibitory functions, additional mechanisms by which HCMV modulates the class I pathway of antigen presentation are likely to exist. Given the very large HCMV proteome, extensive collections of HCMV deletion mutants are instrumental for identifying unrecognized interfering factors and pinpointing responsible HCMV genes.
Observations made in overexpression systems of single proteins suggest further mechanisms of interference with MHC I antigen presentation. The US6 family member US10 was shown to co-precipitate with classical MHC I and to delay the egress of MHC I from the ER in transfected cells.93 However, it is not clear if there is a preference for a specific allele and whether the interaction with US10 could impact the MHC I molecule. US6 family member US8 was found to bind to MHC I heavy chains in the ER, but no effect on MHC I maturation rate or surface disposition was observed.94
The HCMV encoded MHC I homolog UL18 was proposed to interact with TAP, uncoupling the US6-mediated block upon the overexpression of the viral proteins by recombinant vaccinia viruses.95 In this way, UL18 could gain access to peptide ligands and simultaneously interrupt MHC I interaction with the PLC. It was suggested that quality control of MHC I peptide loading is reduced in this manner.95
In HCMV-infected fibroblasts, alternative splicing of the tapasin transcript resulted in an induced level of a tapasin variant lacking exon 3.96 The tapasin variant had lost the ability to interact directly with MHC I and ERp57, but stabilized TAP expression.96 Although protein synthesis of the shorter splice variant has not yet been verified in fibroblasts, the finding exemplifies that alternative splicing induced by HCMV is an additional potential mechanism by which cellular functions could be manipulated.
The HCMV tegument protein pp71 (UL82) was reported to delay MHC I maturation.97 However, to date, no independent studies were able to confirm this effect on MHC I (Ref. 98 and our observations). Further studies are also required to shed light on the expression and function of miR-US4-1. The expression of the sequence99 used to demonstrate that miR-US4-1 downregulates the trimming aminopeptidase ERAP1100 was not verified during infection with the HCMV strain Towne, which shares genomic sequences with the AD169 and Merlin strains in this region.101
HCMV interference with non-classical MHC I molecules
HLA-E and HLA-G are non-classical but peptide-dependent MHC I molecules encoded in the MHC I locus with low levels of heterogeneity compared to classical MHC I molecules. Whereas HLA-E is IFN-γ-inducible, HLA-G with its polymorphic promoter has lost the ability to respond directly to NF-κB and IFN-γ.102 Instead, HLA-G expression can be induced by interferon-inducible transcription factors, such as IRF-1, and other stimuli.103
HLA-E
HLA-E is highly stringent regarding its peptide ligands, with leader sequences from certain HLA-A, -B, -C and -G alleles being its natural ligands.104 Under stress conditions, a leader sequence derived from the 60 kDa heat-shock protein can also be loaded.105 Moreover, in TAP-deficient cells where MHC I leader sequences are lost, HLA-A2-like peptide ligands were found loaded onto HLA-E.106 Peptide bound HLA-E is expressed at low levels on the cell surface, where it serves as a dominant inhibitory ligand for the dimeric CD94/NKG2A (and NKG2B) receptor on NK cells. Whereas the CD94 chain is invariant, NKG2 is variable and the variants C, E and H forward activating signals after engagement with HLA-E.107,108 Thus, it is not surprising that isolated expression of US2 and US11 does not affect HLA-E, while TAP inactivation by US6 clearly impairs HLA-E densities on the plasma membrane.47
HCMV has taken advantage of the very restricted specificity of HLA-E ligands and copied a stretch of amino acids in the UL40 signal peptide that is identical to the HLA-C leader sequence VMAPRTLIL (Figure 3). However, in contrast to MHC I leader sequences, loading of the UL40 peptide onto HLA-E is TAP-independent.109 Although the mechanism by which HCMV conducts the loading is not completely clear, a recent study demonstrated that the positioning of the ligand sequence between two hydrophobic regions at the C-terminus of the long UL40 signal peptide is an important feature for TAP independent loading.84 This assures a peptide supply for HLA-E even in the presence of the simultaneously expressed TAP inhibitor US6.85 Several studies have shown that the expression of UL40 and stabilization of HLA-E exerts a repressive effect on NK cells9,109,110 and that IFN-γ-induced HLA-E expression increases the inhibitory effect on NK cells through UL40.111

HCMV-encoded inhibitors of non-classical MHC I molecules. Inhibition of TAP-dependent peptide translocation by US6 reduces maturation of HLA-E and UL18. To counteract this susceptibility of infected cells to NK cell attack, the signal peptide of UL40 (kinked arrow) provides peptide ligands to HLA-E and UL18 in a TAP-independent manner. Entry of MICB and ULBP1, ULBP2 and ULBP6 into the secretory pathway is blocked by UL16, whereas UL142 blocks MICA and ULBP3. US18 and US20 direct MICA for degradation in lysosomal compartments. HLA-G is directed for proteasomal degradation by both US2 and US10. HCMV, human cytomegalovirus; MHC, major histocompatibility complex; MICA, MHC class I chain-related A; NK, natural killer; ULBP, UL16-binding protein.
Interestingly, HLA-E reactive NKT (NK-CTL) cells were found in HCMV-positive individuals who do not have HLA alleles that contain the VMAPRTLIL sequence. Consequently, tolerance towards the VMAPRTLIL-HLA-E complexes is not present and HLA-E presentation of the UL40-derived peptide was shown to induce CTL reactivity.112,113,114 Sequencing of a number of HCMV clinical strains revealed that even though VMAPRTLIL represents the most common sequence, variable sequences also exist within the UL40 signal peptide.84,115,116 A detailed analysis of the propensities of such UL40 variations showed that they have different impacts on the affinity for the inhibitory NKG2A and activating NKG2C receptors when loaded onto HLA-E. Some even abolish the ability to stabilize HLA-E molecules.116 It is conceivable that variations in the UL40 sequence become selected when the virus is confronted with HLA-E reactive NK-CTL cells.
A significant expansion of CD94/NKG2C-positive NK cells was consistently observed in HCMV-positive subjects.17 Cocultivation of PBMCs with HCMV-infected fibroblasts reproduced the expansion of this NK cell subset in vitro.117 It is not clear whether HLA-E expression on HCMV-infected cells is required for the expansion of this NK cell subpopulation. Cocultivation of PBMCs using a UL40 deletion mutant of HCMV did not have an effect on CD94/NKG2C+ cell expansion, suggesting that a mechanism other than the upregulation of HLA-E is required.117 Moreover, when a HCMV mutant was applied that lacked the gene region US2-US11 encompassing the established MHC I inhibitors, the expansion of CD94/NKG2C+ cells was blocked.117 This finding implies that MHC I expression prohibits the expansion of these NK cells, possibly by engaging KIR receptors. However, it has still not been excluded that an NK cell ligand stimulating the expansion of CD94/NKG2C+ NK cells is encoded in this region.
Intriguingly, a recent study118 on miRNA regulation of HCMV-infected cells adds a twist to the sophisticated viral HLA-E control. HCMV infection induces the expression of the RNA editing protein ADAR p110, which leads to increased levels of the edited form of miR-376a. The edited miRNA specifically targets HLA-E transcripts and reduces HLA-E biosynthesis, resulting in induced NK cell recognition of infected cells. The effect on NK cells was dependent on UL40-induced HLA-E expression,118 meaning that HCMV stabilizes HLA-E expression by UL40 and simultaneously reduces the translation of HLA-E by the upregulation of ADAR p110 and edited miR-376a. The expression kinetics of UL40 and ADAR p110 significantly overlap (E), highlighting the delicate balance of both effects.
HLA-G
HLA-G expression was first thought to be restricted to fetal trophoblasts regulating NK cells at the fetal–maternal interface during pregnancy. Compelling data linking HLA-G to immune inhibition and tolerance has given HLA-G additional attention (reviewed in Ref. 119). In its homodimeric form, HLA-G binds to KIR2DL4, LIR-1 and LIR-2 with a higher affinity than classical MHC I molecules, and therefore functions as a potent inhibitory ligand.120 Because LIR-1 and LIR-2 are expressed not only on subsets of NK cells, but also on monocytes and macrophages, and, in addition, LIR-1 is expressed on B cells and some CD4+ and CD8+ T-cell subsets, HLA-G is now considered to function as an important immunomodulatory molecule. Indeed, the expression of HLA-G on myeloid cells can be regulated by cytokines, such as IL-10 and interferons.121,122
Seven differently spliced transcripts from the HLA-G gene have been described.119 This reorganization of exons results in different combinations of the α1-3 domains with and without a TMS and dimerization with β2m. The HLA-G1 transcript corresponds to the full length type I transmembrane protein and is the most abundant transcript, but HLA-G5, which encodes for a shorter soluble form of HLA-G1 lacking a TMS but still capable of dimer formation with β2m, is expressed by myeloid cells.
It has been reported that HLA-G is targeted by US2 resulting in its degradation44 (Figure 3). Furthermore, it is interesting that while US10 interacts with classical MHC I and reduces their maturation rate,93 HLA-G is destabilized by US10 in a proteasome-dependent manner.123 A tri-leucine motif in the cytoplasmic tail of US10 was found to be crucial for HLA-G degradation. The C-terminal tail of HLA-G was important for this function, while the tail of HLA-A2 blocked US10-mediated degradation.123 This suggests that US10 can only target transcript variants of HLA-G with a cytosolic tail if the direct contact via the ectodomains remains intact. For understanding of the respective role that US2- and US10-mediated HLA-G degradation plays in HCMV immune control requires further investigation in the future.
NKG2D ligands: MICA, MICB and ULBP1–6
The NKG2D ligands are broadly expressed stress-induced molecules that trigger the activating CD94/NKG2D receptor, which is expressed on most NK cells and a substantial subpopulation of CD8+ T cells. MICA and MICB are type I transmembrane proteins with preserved α1-3 domains that do not form dimers with β2m. MICA and MICB are encoded in the MHC locus and are highly polymorphic, with 79 and 26 different proteins currently reported, respectively (according to the IMGT/HLA database June 2014).
The ULBP family of ligands is distinct in that they lack an α3 domain and TMS. Instead, they are attached to the membrane via a glycosyl-phosphatidylinositol anchor (except for ULBP4, which is a predicted type I transmembrane protein124). MICA, MICB and the ULBPs do not require a peptide ligand for conformational stabilization. The expression of the NKG2D ligands is non-constitutive and still ill-defined, but by possibly diverse stress signals, their expression can be strongly induced in a wide variety of cells. HCMV infection efficiently induces the expression of NKG2D ligands with the exception of ULBP4.125 The HCMV pp86 IE2 protein was found to induce more potent upregulation of MICA and MICB, whereas IE1 induced the expression of ULBP2,126 emphasizing the different pathways of induction for these ligands.
UL16 was the first HCMV encoded protein that was found to interfere with the expression of the NKG2D ligands (Figure 3). UL16 binds and inhibits cell surface expression of MICB and ULBP1–2 and 6.127,128,129,130 This interaction leads to retention and accumulation of Endoglycosidase H-sensitive forms of MICB and is dependent on the cytoplasmic tail of UL16.129 The expression of a UL16 mutant lacking a tyrosine-based motif in the cytoplasmic tail was not able to inhibit surface expression of MICB.131 Domain swapping experiments revealed that the α2 domain of MICB was sufficient for UL16 to target MICA/B chimeras.132 Indeed, the crystal structure of a UL16–MICB complex revealed that several residues in the α2 domain are contacted by UL16.133 Interestingly, despite lacking sequence homologies, by convergent evolution, UL16 has adapted an interaction mode with MICB highly similar to the one used by NKG2D.133 In the center of the complex, UL16 contacts the α3-helix of MICB and forms a saddle and horseback-like structure. Based on the structure, it was suggested that exchange of glutamine 169 to arginine (as in MICA and ULBP3) may prevent binding by UL16 and therefore, could explain the specificity of UL16 for MICB and ULBP1–2 and 6. This is also in agreement with mutational analysis of UL16 binding to MICB.132
The selective pressure by UL16 possibly led to the emergence of NKG2D ligand variants. It is conceivable to assume that HCMV-encoded inhibitors have resulted in the diversification of MICA, MICB and ULBPs. One example is the MICA allele MICA*008, which has gained a premature stop codon, resulting in loss of the TMS. MICA*008 is anchored to the membrane by glycosyl-phosphatidylinositol. This mutation rendered the allele MICA*008 resistant to UL142-mediated inhibition of type I transmembrane MICA proteins.134
UL142 is a MHC I-like protein that inhibits the expression of MICA and ULBP3.135,136 Similar to UL16, UL142-targeted proteins are not degraded but retained in an intracellular compartment, in this case, in the ER and cis-Golgi (Figure 3).135,136 It was shown that the TMS of UL142 is decisive for ER retention and that both the luminal domain and the TMS are involved in the retention of MICA.135
Recently, new HCMV-encoded inhibitors of MICA have been identified; US18 and US20 belong to the still little characterized US12 gene family encoding seven-transmembrane domain proteins (Figure 3). US18 and US20 redirect MICA to lysosomal compartments for proteolysis, while they exert no effect on MICB and ULBP2.126
Ligands of NKG2D are not only targeted by HCMV proteins. MICB expression is selectively blocked by one of the HCMV-encoded miRNAs (miR-UL112).137 This additional level of control highlights the arms race between NKG2D ligands of the host and HCMV countermeasures. The multitude of variable inhibitory factors (including UL16, a type I transmembrane protein; UL142, a MHC I-like protein; US18 and US20, seven-transmembrane domain proteins; and miR-UL112, a miRNA) reflects the dominant selective pressure exerted by a single receptor (i.e., CD94/NKG2D) in controlling HCMV replication.
UL18
Due to its sequence homology with MHC I proteins, the UL18 molecule gained immediate attention and was the first immunoevasin identified in HCMV.26,138 Because of its ability to form a tight complex with β2m, it was surmised to be an inhibitor of CD8+ T-cell recognition by sequestering β2m and thus impairing MHC I formation.26 Although impaired levels of MHC I assembly in HCMV-infected cells were documented, this idea was discarded due to unchanged levels of free β2m and a lack of UL18-dependent effects on MHC I molecules.139 The discovery of peptide binding by UL18 strengthened the idea that UL18 could act as a ligand for NK cells.140
A screen for UL18 binding partners led to the discovery of LIR-16. LIR-1 is an MHC I receptor expressed on various cells, including monocytes, dendritic cells, B cells and a subset of NK cells and T-cells. LIR-1 has now been shown to bind to a broad range of MHC I molecules.6,7,8 The affinity of LIR-1 for UL18 is 1000-fold stronger than for HLA-A2,141 and might therefore allow for an efficient interaction despite the low surface expression of UL18.142
Although clinical strains exhibit a certain degree of sequence variability in the UL18 gene, all variants are able to bind LIR-1, supporting the idea that this interaction is critical for UL18 function.143,144 Additional MHC-like molecules have been proposed to be encoded by the genes UL142 and UL37, respectively.145,146 Crystal structures for these proteins are not yet available and no function has been assigned for UL37. To date, of the determined viral MHC I-like structures, UL18 is the only molecule that binds peptides.147 Although the UL18 ectodomain shares only approximately 25% homology with classical MHC I molecules,138 it folds with high similarly to HLA-A2 (Figure 4a and b).148 It is fascinating that whereas human MHC I molecules carry only one N-linked glycosylation site, UL18 has gained 13 potential glycosylation sites (sites are depicted in pink in Figure 4b and c), a majority of which are used. These glycans are not important for UL18 binding to LIR-1,148 but might block possible interactions with other MHC I binding proteins, such as the TCR, CD8 and KIRs,148 thereby prohibiting the initiation of undesired immune reactions. The glycans might also be a reason why UL18 fully escapes the viral inhibitors US2, US3 and US11,149 while preserving the accessibility for its true receptor, the inhibitory LIR-1.

HLA-A*02 and UL18 in complex with LIR-1. (a) Crystal structure of HLA-A*02 bound to LIR-1.5 The HLA-A*02 chain is shown in turquoise, β2m in yellow and LIR-1 domains D1-2 in green. (b) UL18 is shown in blue148 with putative glycosylated asparagines highlighted in pink. (c) The UL18–LIR-1 structure from B turned horizontally by 180°. LIR, leukocyte Ig-like receptor.
LIR-1 is a type I transmembrane protein consisting of four Ig-like domains (D1–4). The cytosolic tail harbors four ITIM repeats150 and therefore classifies LIR-1 as an inhibitory receptor. Nevertheless, the verification of the anticipated role of UL18 in the inhibition of NK cell activation through engagement with the LIR-1 receptor was slow in coming due to contradictory observations. Whereas Reyburn et al.151 reported NK cell inhibition after the ectopic expression of UL18,151 this was not observed by Leong et al.142 In the latter work, both the usage of an HCMV UL18 deletion mutant and ectopic UL18 expression indicated that the presence of UL18 induced NK cell responses. One attempt to solve this controversy was made by Prod'homme et al.,152 who found segregating effects, including the activation of LIR-1− NK cells and inhibition of LIR-1+ NK cells. The activation of LIR-1− NK cells could be a consequence of weak UL18 binding to CD94/NKG2C.153
Because LIR-1 is only expressed by a subset of NK cells, it is unclear whether NK cells are the main target population or whether additional cell types are also influenced by UL18. Indeed, a UL18 fusion protein impacted dendritic cell maturation, motility and cytokine production.154 It should be noted that whereas a UL18 fusion protein lowered IFN-γ production by PBMC after coculture with HCMV-infected cells, UL18 expressed in the context of HCMV infection induced IFN-γ production,155 emphasizing the delicate balance of receptor triggering and the resulting immunological responses. This indeed calls for precaution in evaluating various expression systems. Along this line, the overexpression of UL18 by adenoviral or vaccinia virus vectors has proven to more efficiently display UL18 on the cell surface than expression in the context of HCMV infection. Nonetheless, UL18 is detected on the surface of HCMV-infected fibroblasts at late time-points of infection and the expression density increases with proceeding of the replication cycle.156 This is possibly part of a more general strategy of HCMV: exposure of potential antigens on the cell surface is only allowed at late time points of infection to minimize antibody-dependent effector functions during the early stages of the replication cycle.157 The kinetics of UL18 surface expression were shown to be regulated by two motifs in the cytoplasmic tail of UL18, causing ER retention and internalization.156 Indeed, it has been suggested that UL18 might confer both intracellular and extracellular functions,155 which could be segregated in a manner dependent on the phase of HCMV replication.
What can we expect from future studies?
Progressive expression of HCMV-encoded MHC I inhibitors results in the continuous reduction of MHC I expression at the cell surface, with an almost complete downregulation when infected fibroblasts are analyzed at the end of the replication cycle in vitro. Simultaneously, the expression of multiple NK cell inhibitors accelerates, being in its full-blown state, when the MHC I molecule density is very low.158 These coordinated effects tell us that the level of MHC I down-regulation aiming at CD8+ T-cell evasion causes high immunological costs which must be compensated for by extensive genomic investments in factors that rescue the vulnerable HCMV-infected cell from NK cell recognition. Despite numerous HCMV NK cell inhibitors defined to date, it is tempting to speculate that the absolute number of HCMV factors interfering with NK cell functions is still far from complete.
A frequently discussed issue is the high number and functional redundancy of immunoevasins that are a typical feature of cytomegalovirus counteraction against immune responses,as highlighted with regard to CD8+ T cells, NK cells, IgG and IFN. Only the high number of immunoevasive genes may put the virus in a position to respond both most effectively and flexibly to widely varying host conditions. Cytokines, such as IFN-γ, IFN-α or TNF-α, which upregulate MHC expression, were shown to influence the extent of surface resident MHC I and antigen presentation to CD8+ T cells following HCMV infection.31,159 What has attracted our attention only recently, is the specific role the HCMV-infected target cell type will have. During the infection of its host, HCMV replicates in a large variety of cells, including endothelial cells, epithelial cells and myeloid cells.160 Still, most studies were carried out using HCMV-permissive fibroblasts that likely provide optimal conditions for the immunological effectiveness of immunoevasins, while infected macrophages or dendritic cells are readily recognized by both CD8+ and NK cells.161,162,163 It is conceivable to assume that HCMV benefits from the sensitization of infected DCs for NK cell destruction161 due to subsequently impaired activation of T-cell responses.
In conclusion, more than two decades of research has provided an impressive body of evidence that HCMV has learned to pay much attention to all aspects of classical and non-classical MHC class I molecules. However, limited knowledge exists regarding whether the virus can also control peptide ligand processing and MHC I peptide selection and loading. Considering the high immunological costs incurred from non-selective MHC I molecule downregulation as a strategy for CD8+ T-cell escape, MHC ligand-specific effects could offer a way out. Future insight into the HCMV ligandome may further increase our understanding of MHC I manipulation.
References
Ljunggren HG, Karre K . In search of the ‘missing self': MHC molecules and NK cell recognition. Immunol Today 1990; 11: 237–244.
Parham P, Moffett A . Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol 2013; 13: 133–144.
Blais ME, Dong T, Rowland-Jones S . HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? Immunology 2011; 133: 1–7.
Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ, Stuart DI et al. Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature 1997; 387: 630–634.
Willcox BE, Thomas LM, Bjorkman PJ . Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol 2003; 4: 913–919.
Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 1997; 7: 273–282.
Lepin EJ, Bastin JM, Allan DS, Roncador G, Braud VM, Mason DY et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol 2000; 30: 3552–3561.
Navarro F, Llano M, Bellón T, Colonna M, Geraghty DE, López-Botet M . The ILT2(LIR1) and CD94/NKG2A NK cell receptors respectively recognize HLA-G1 and HLA-E molecules co-expressed on target cells. Eur J Immunol 1999; 29: 277–283.
Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD . Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 2000; 405: 537–543.
Hennecke J, Wiley DC . T cell receptor-MHC interactions up close. Cell 2001; 104: 1–4.
Champsaur M, Lanier LL . Effect of NKG2D ligand expression on host immune responses. Immunol Rev 2010; 235: 267–285.
Mocarski ES, Shenk T, Pass RF . Cytomegaloviruses. In: Knipe DK, Howley PM (ed.) Field's Virology. Philadelphia, PA: Lippincott, Williams & Wilkins, 2007: 2701–2772.
Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX et al. Decoding human cytomegalovirus. Science 2012; 338: 1088–1093.
Ludwig A, Hengel H . Epidemiological impact and disease burden of congenital cytomegalovirus infection in Europe. Euro Surveill 2009; 14: 26–32.
Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 2005; 202: 673–685.
Beziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Björklund AT et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013; 121: 2678–2688.
Guma M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M . Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004; 104: 3664–3671.
Koch J, Guntrum R, Heintke S, Kyritsis C, Tampe R . Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J Biol Chem 2004; 279: 10142–10147.
Hulpke S, Tomioka M, Kremmer E, Ueda K, Abele R, Tampé R . Direct evidence that the N-terminal extensions of the TAP complex act as autonomous interaction scaffolds for the assembly of the MHC I peptide-loading complex. Cell Mol Life Sci 2012; 69: 3317–3327.
Panter MS, Jain A, Leonhardt RM, Ha T, Cresswell P . Dynamics of major histocompatibility complex class I association with the human peptide-loading complex. J Biol Chem 2012; 287: 31172–31184.
Lehner PJ, Surman MJ, Cresswell P . Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line.220. Immunity 1998; 8: 221–231.
Tan P, Kropshofer H, Mandelboim O, Bulbuc N, Hämmerling GJ, Momburg F . Recruitment of MHC class I molecules by tapasin into the transporter associated with antigen processing-associated complex is essential for optimal peptide loading. J Immunol 2002; 168: 1950–1960.
Chang SC, Momburg F, Bhutani N, Goldberg AL . The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc Natl Acad Sci USA 2005; 102: 17107–17112.
Wearsch PA, Cresswell P . Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat Immunol 2007; 8, 873–881.
Roder G, Geironson L, Rasmussen M, Harndahl M, Buus S, Paulsson K . Tapasin discriminates peptide-human leukocyte antigen-A*02:01 complexes formed with natural ligands. J Biol Chem 2011; 286: 20547–20557.
Browne H, Smith G, Beck S, Minson T . A complex between the MHC class I homologue encoded by human cytomegalovirus and beta 2 microglobulin. Nature 1990; 347, 770–772.
Barnes PD, Grundy JE . Down-regulation of the class I HLA heterodimer and beta 2-microglobulin on the surface of cells infected with cytomegalovirus. J Gen Virol 1992; 73( Pt 9): 2395–2403.
Yamashita Y, Shimokata K, Mizuno S, Yamaguchi H, Nishiyama Y . Down-regulation of the surface expression of class I MHC antigens by human cytomegalovirus. Virology 1993; 193: 727–736.
Beersma MF, Bijlmakers MJ, Ploegh HL . Human cytomegalovirus down-regulates HLA class I expression by reducing the stability of class I H chains. J Immunol 1993; 151: 4455–4464.
Warren AP, Ducroq DH, Lehner PJ, Borysiewicz LK . Human cytomegalovirus-infected cells have unstable assembly of major histocompatibility complex class I complexes and are resistant to lysis by cytotoxic T lymphocytes. J Virol 1994; 68: 2822–2829.
Hengel H, Esslinger C, Pool J, Goulmy E, Koszinowski UH . Cytokines restore MHC class I complex formation and control antigen presentation in human cytomegalovirus-infected cells. J Gen Virol 1995; 76( Pt 12): 2987–2997.
Hengel H, Flohr T, Hammerling GJ, Koszinowski UH, Momburg F . Human cytomegalovirus inhibits peptide translocation into the endoplasmic reticulum for MHC class I assembly. J Gen Virol 1996; 77( Pt 9): 2287–2296.
Reddehase MJ . Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol 2002; 2: 831–844.
Jones TR, Sun L . Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. J Virol 1997; 71: 2970–2979.
Jones TR, Hanson LK, Sun L, Slater JS, Stenberg RM, Campbell AE Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J Virol 1995; 69: 4830–4841.
Jones TR, Wiertz EJ, Sun L, Fish KN, Nelson JA, Ploegh HL . Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc Natl Acad Sci USA 1996; 93: 11327–11333.
Tenney DJ, Colberg-Poley AM . Human cytomegalovirus UL36–38 and US3 immediate-early genes: temporally regulated expression of nuclear, cytoplasmic, and polysome-associated transcripts during infection. J Virol 1991; 65: 6724–6734.
Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJ, Ploegh HL et al. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997; 6: 613–621.
Hengel H, Koopmann JO, Flohr T, Muranyi W, Goulmy E, Hämmerling GJ et al. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 1997; 6: 623–632.
Lehner PJ, Karttunen JT, Wilkinson GW, Cresswell P . The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc Natl Acad Sci USA 1997; 94: 6904–6909.
Gewurz BE, Gaudet R, Tortorella D, Wang EW, Ploegh HL, Wiley DC et al. Antigen presentation subverted: structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc Natl Acad Sci USA 2001; 98: 6794–6799.
Gewurz BE, Wang EW, Tortorella D, Schust DJ, Ploegh HL . Human cytomegalovirus US2 endoplasmic reticulum-lumenal domain dictates association with major histocompatibility complex class I in a locus-specific manner. J Virol 2001; 75: 5197–5204.
Gewurz BE, Ploegh HL, Tortorella D . US2, a human cytomegalovirus-encoded type I membrane protein, contains a non-cleavable amino-terminal signal peptide. J Biol Chem 2002; 277: 11306–11313.
Barel MT, Ressing M, Pizzato N, van Leeuwen D, Le Bouteiller P, Lenfant F et al. Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J Immunol 2003; 171: 6757–6765.
Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch K M. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity 2009; 30: 21–32.
Barel MT, Hassink GC, van Voorden S, Wiertz EJ . Human cytomegalovirus-encoded US2 and US11 target unassembled MHC class I heavy chains for degradation. Mol Immunol 2006; 43: 1258–1266.
Llano M, Guma M, Ortega M, Angulo A, Lopez-Botet M . Differential effects of US2, US6 and US11 human cytomegalovirus proteins on HLA class Ia and HLA-E expression: impact on target susceptibility to NK cell subsets. Eur J Immunol 2003; 33: 2744–2754.
Ameres S, Besold K, Plachter B, Moosmann A . CD8 T cell-evasive functions of human cytomegalovirus display pervasive MHC allele specificity, complementarity, and cooperativity. J Immunol 2014; in press.
Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR et al. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996; 384: 432–438.
Hassink GC, Barel MT, van Voorden SB, Kikkert M, Wiertz EJ . Ubiquitination of MHC class I heavy chains is essential for dislocation by human cytomegalovirus-encoded US2 but not US11. J Biol Chem 2006; 281: 30063–30071.
Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, Gemmill RM et al. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol 2009; 186: 685–692S.
Loureiro J, Lilley BN, Spooner E, Noriega V, Tortorella D, Ploegh HL . Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature 2006; 441: 894–897.
Lee SO, Cho K, Cho S, Kim I, Oh C, Ahn K . Protein disulphide isomerase is required for signal peptide peptidase-mediated protein degradation. EMBO J 2010; 29: 363–375.
Ben-Arieh SV, Zimerman B, Smorodinsky NI, Yaacubovicz M, Schechter C, Bacik I et al. Human cytomegalovirus protein US2 interferes with the expression of human HFE, a nonclassical class I major histocompatibility complex molecule that regulates iron homeostasis. J Virol 2001; 75: 10557–10562.
Vahdati-Ben Arieh S, Laham N, Schechter C, Yewdell JW, Coligan JE, Ehrlich R . A single viral protein HCMV US2 affects antigen presentation and intracellular iron homeostasis by degradation of classical HLA class I and HFE molecules. Blood 2003; 101: 2858–2864.
Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR et al. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 1999; 5: 1039–1043.
Han J, Rho SB, Lee JY, Bae J, Park SH, Lee SJ et al. Human cytomegalovirus (HCMV) US2 protein interacts with human CD1d (hCD1d) and down-regulates invariant NKT (iNKT) cell activity. Mol Cells 2013; 36: 455–464.
Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL . The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996; 84: 769–779.
Lilley BN, Ploegh HL . A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004; 429: 834–840.
Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA . A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004; 429: 841–847.
Lee SO, Hwang S, Park J, Park B, Jin BS, Lee S et al. Functional dissection of HCMV US11 in mediating the degradation of MHC class I molecules. Biochem Biophys Res Commun 2005; 330: 1262–1267.
Lilley BN, Tortorella D, Ploegh HL . Dislocation of a type I membrane protein requires interactions between membrane-spanning segments within the lipid bilayer. Mol Biol Cell 2003; 14: 3690–3698.
Barel MT, Pizzato N, van Leeuwen D, Bouteiller PL, Wiertz EJ, Lenfant F . Amino acid composition of alpha1/alpha2 domains and cytoplasmic tail of MHC class I molecules determine their susceptibility to human cytomegalovirus US11-mediated down-regulation. Eur J Immunol 2003; 33: 1707–1716.
Cho S, Kim BY, Ahn K, Jun Y . The C-terminal amino acid of the MHC-I heavy chain is critical for binding to Derlin-1 in human cytomegalovirus US11-induced MHC-I degradation. PLoS One 2013; 8: e72356.
Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA . Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 2005; 102: 14132–14138.
Mueller B, Lilley BN, Ploegh HL . SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol 2006; 175: 261–270.
van de Weijer ML, Bassik MC, Luteijn RD, Voorburg CM, Lohuis MA, Kremmer E et al. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat Commun 2014; 5: 3832.
van den Boomen DJ, Timms RT, Grice GL, Stagg HR, Skødt K, Dougan G et al. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc Natl Acad Sci USA 2014; 111: 11425–11430.
Flierman D, Coleman CS, Pickart CM, Rapoport TA, Chau V . E2-25K mediates US11-triggered retro-translocation of MHC class I heavy chains in a permeabilized cell system. Proc Natl Acad Sci USA 2006; 103: 11589–11594.
Story CM, Furman MH, Ploegh HL . The cytosolic tail of class I MHC heavy chain is required for its dislocation by the human cytomegalovirus US2 and US11 gene products. Proc Natl Acad Sci USA 1999; 96: 8516–8521.
Schust DJ, Tortorella D, Seebach J, Phan C, Ploegh HL . Trophoblast class I major histocompatibility complex (MHC) products are resistant to rapid degradation imposed by the human cytomegalovirus (HCMV) gene products US2 and US11. J Exp Med 1998; 188: 497–503.
Ameres S, Mautner J, Schlott F, Neuenhahn M, Busch DH, Plachter B et al. Presentation of an immunodominant immediate-early CD8+ T cell epitope resists human cytomegalovirus immunoevasion. PLoS Pathog 2013; 9: e1003383.
Cho S, Lee M, Jun Y . Forced interaction of cell surface proteins with Derlin-1 in the endoplasmic reticulum is sufficient to induce their dislocation into the cytosol for degradation. Biochem Biophys Res Commun 2013; 430: 787–792.
Gruhler A, Peterson PA, Fruh K . Human cytomegalovirus immediate early glycoprotein US3 retains MHC class I molecules by transient association. Traffic 2000; 1: 318–325.
Huard B, Fruh K . A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur J Immunol 2000; 30: 509–515.
Park B, Kim Y, Shin J, Lee S, Cho K, Früh K et al. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 2004; 20: 71–85.
Noriega VM, Hesse J, Gardner TJ, Besold K, Plachter B, Tortorella D . Human cytomegalovirus US3 modulates destruction of MHC class I molecules. Mol Immunol 2012; 51: 245–253.
Misaghi S, Sun ZY, Stern P, Gaudet R, Wagner G, Ploegh H . Structural and functional analysis of human cytomegalovirus US3 protein. J Virol 2004; 78: 413–423.
Lee S, Park B, Ahn K . Determinant for endoplasmic reticulum retention in the luminal domain of the human cytomegalovirus US3 glycoprotein. J Virol 2003; 77: 2147–2156.
Lee S, Yoon J, Park B, Jun Y, Jin M, Sung HC et al. Structural and functional dissection of human cytomegalovirus US3 in binding major histocompatibility complex class I molecules. J Virol 2000; 74: 11262–11269.
Shin J, Park B, Lee S, Kim Y, Biegalke BJ, Kang S et al. A short isoform of human cytomegalovirus US3 functions as a dominant negative inhibitor of the full-length form. J Virol 2006; 80: 5397–5404.
Jun Y, Kim E, Jin M, Sung HC, Han H, Geraghty DE et al. Human cytomegalovirus gene products US3 and US6 down-regulate trophoblast class I MHC molecules. J Immunol 2000; 164: 805–811.
Wei ML, Cresswell P . HLA-A2 molecules in an antigen-processing mutant cell contain signal sequence-derived peptides. Nature 1992; 356: 443–446.
Prod'homme V, Tomasec P, Cunningham C, Lemberg MK, Stanton RJ, McSharry BP et al. Human cytomegalovirus UL40 signal peptide regulates cell surface expression of the NK cell ligands HLA-E and gpUL18. J Immunol 2012; 188: 2794–2804.
Ulbrecht M, Hofmeister V, Yüksekdag G, Ellwart JW, Hengel H, Momburg F et al. HCMV glycoprotein US6 mediated inhibition of TAP does not affect HLA-E dependent protection of K-562 cells from NK cell lysis. Hum Immunol 2003; 64: 231–237.
Gorbulev S, Abele R, Tampe R . Allosteric crosstalk between peptide-binding, transport, and ATP hydrolysis of the ABC transporter TAP. Proc Natl Acad Sci USA 2001; 98: 3732–3737.
Hewitt EW, Gupta SS, Lehner PJ . The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J 2001; 20: 387–396.
Verweij MC, Ressing ME, Knetsch W, Quinten E, Halenius A, van Bel N et al. Inhibition of mouse TAP by immune evasion molecules encoded by non-murine herpesviruses. Mol Immunol 2011; 48: 835–845.
Halenius A, Momburg F, Reinhard H, Bauer D, Lobigs M, Hengel H . Physical and functional interactions of the cytomegalovirus US6 glycoprotein with the transporter associated with antigen processing. J Biol Chem 2006; 281: 5383–5390.
Dugan GE, Hewitt EW . Structural and functional dissection of the human cytomegalovirus immune evasion protein US6. J Virol 2008; 82: 3271–3282.
Powers CJ, Fruh K . Signal peptide-dependent inhibition of MHC class I heavy chain translation by rhesus cytomegalovirus. PLoS Pathog 2008; 4: e1000150.
Halenius A, Hauka S, Dölken L, Stindt J, Reinhard H, Wiek C et al. Human cytomegalovirus disrupts the major histocompatibility complex class I peptide-loading complex and inhibits tapasin gene transcription. J Virol 2011; 85: 3473–3485.
Furman MH, Dey N, Tortorella D, Ploegh HL . The human cytomegalovirus US10 gene product delays trafficking of major histocompatibility complex class I molecules. J Virol 2002; 76: 11753–11756.
Tirabassi RS, Ploegh HL . The human cytomegalovirus US8 glycoprotein binds to major histocompatibility complex class I products. J Virol 2002; 76: 6832–6835.
Kim Y, Park B, Cho S, Shin J, Cho K, Jun Y et al. Human cytomegalovirus UL18 utilizes US6 for evading the NK and T-cell responses. PLoS Pathog 2008; 4: e1000123.
Beutler N, Hauka S, Niepel A, Kowalewski DJ, Uhlmann J, Ghanem E et al. A natural tapasin isoform lacking exon 3 modifies peptide loading complex function. Eur J Immunol 2013; 43: 1459–1469.
Trgovcich J, Cebulla C, Zimmerman P, Sedmak DD . Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J Virol 2006; 80: 951–963.
Hesse J, Reyda S, Tenzer S, Besold K, Reuter N, Krauter S et al. Human cytomegalovirus pp71 stimulates major histocompatibility complex class I presentation of IE1-derived peptides at immediate early times of infection. J Virol 2013; 87: 5229–5238.
Grey F, Antoniewicz A, Allen E, Saugstad J, McShea A, Carrington JC et al. Identification and characterization of human cytomegalovirus-encoded microRNAs. J Virol 2005; 79: 12095–12099.
Kim S, Lee S, Shin J, Kim Y, Evnouchidou I, Kim D et al. Human cytomegalovirus microRNA miR-US4-1 inhibits CD8+ T cell responses by targeting the aminopeptidase ERAP1. Nat Immunol 2011; 12: 984–991.
Stark TJ, Arnold JD, Spector DH, Yeo GW . High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J Virol 2012; 86: 226–235.
Chu W, Gao J, Murphy WJ, Hunt JS . A candidate interferon-gamma activated site (GAS element) in the HLA-G promoter does not bind nuclear proteins. Hum Immunol 1999; 60: 1113–1118.
Alegre E, Rizzo R, Bortolotti D, Fernandez-Landázuri S, Fainardi E, González A Some basic aspects of HLA-G biology. J Immunol Res 2014; 2014: 657625.
Braud V, Jones EY, McMichael A . The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol 1997; 27: 1164–1169.
Michaelsson J, Teixeira de Matos C, Achour A, Lanier LL, Kärre K, Söderström K . A signal peptide derived from hsp60 binds HLA-E and interferes with CD94/NKG2A recognition. J Exp Med 2002; 196: 1403–1414.
Lampen MH, Hassan C, Sluijter M, Geluk A, Dijkman K, Tjon JM et al. Alternative peptide repertoire of HLA-E reveals a binding motif that is strikingly similar to HLA-A2. Mol Immunol 2013; 53: 126–131.
Braud VM, Allan DS, O'Callaghan CA, Söderström K, D'Andrea A, Ogg GS et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998; 391: 795–799.
Kaiser BK, Barahmand-Pour F, Paulsene W, Medley S, Geraghty DE, Strong RK . Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J Immunol 2005; 174: 2878–2884.
Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000; 287: 1031.
Ulbrecht M, Martinozzi S, Grzeschik M, Hengel H, Ellwart JW, Pla M et al. Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J Immunol 2000; 164: 5019–5022.
Cerboni C, Mousavi-Jazi M, Wakiguchi H, Carbone E, Kärre K, Söderström K . Synergistic effect of IFN-gamma and human cytomegalovirus protein UL40 in the HLA-E-dependent protection from NK cell-mediated cytotoxicity. Eur J Immunol 2001; 31: 2926–2935.
Mazzarino P, Romagnani C, Mazzarino P, Falco M, Millo E, Moretta A et al. Identification of effector-memory CMV-specific T lymphocytes that kill CMV-infected target cells in an HLA-E-restricted fashion. Eur J Immunol 2005; 35: 3240–3247.
Pietra G, Romagnani C, Mazzarino P, Falco M, Millo E, Moretta A et al. HLA-E-restricted recognition of cytomegalovirus-derived peptides by human CD8+ cytolytic T lymphocytes. Proc Natl Acad Sci USA 2003; 100: 10896–10901.
Romagnani C, Pietra G, Falco M, Mazzarino P, Moretta L, Mingari MC . HLA-E-restricted recognition of human cytomegalovirus by a subset of cytolytic T lymphocytes. Hum Immunol 2004; 65: 437–445.
Garrigue I, Faure-Della Corte M, Magnin N, Recordon-Pinson P, Couzi L . UL40 human cytomegalovirus variability evolution patterns over time in renal transplant recipients. Transplantation 2008; 86: 826–835.
Heatley SL, Pietra G, Lin J, Widjaja JM, Harpur CM, Lester S et al. Polymorphism in human cytomegalovirus UL40 impacts on recognition of human leukocyte antigen-E (HLA-E) by natural killer cells. J Biol Chem 2013; 288: 8679–8690.
Guma M, Budt M, Sáez A, Brckalo T, Hengel H, Angulo A . Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006; 107: 3624–3631.
Nachmani D, Zimmermann A, Oiknine Djian E, Weisblum Y, Livneh Y, Khanh Le VT et al. MicroRNA editing facilitates immune elimination of HCMV infected cells. PLoS Pathog 2014; 10: e1003963.
Carosella ED, Gregori S, LeMaoult J . The tolerogenic interplay(s) among HLA-G, myeloid APCs, and regulatory cells. Blood 2011; 118: 6499–6505.
Gonen-Gross T, Achdout H, Gazit R, Hanna J, Mizrahi S, Markel G et al. Complexes of HLA-G protein on the cell surface are important for leukocyte Ig-like receptor-1 function. J Immunol 2003; 171: 1343–1351.
Lefebvre S, Moreau P, Guiard V, Ibrahim EC, Adrian-Cabestre F, Menier C et al. Molecular mechanisms controlling constitutive and IFN-gamma-inducible HLA-G expression in various cell types. J Reprod Immunol 1999; 43: 213–224.
Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood 2010; 116: 935–944.
Park B, Spooner E, Houser BL, Strominger JL, Ploegh HL . The HCMV membrane glycoprotein US10 selectively targets HLA-G for degradation. J Exp Med 2010; 207: 2033–2041.
Chalupny NJ, Sutherland CL, Lawrence WA, Rein-Weston A, Cosman D . ULBP4 is a novel ligand for human NKG2D. Biochem Biophys Res Commun 2003; 305: 129–135.
Eagle RA, Traherne JA, Ashiru O, Wills MR, Trowsdale J . Regulation of NKG2D ligand gene expression. Hum Immunol 2006; 67: 159–169.
Fielding CA, Aicheler R, Stanton RJ, Wang EC, Han S, Seirafian S et al. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog 2014; 10: e1004058.
Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001; 14: 123–133.
Vales-Gomez M, Browne H, Reyburn HT . Expression of the UL16 glycoprotein of Human Cytomegalovirus protects the virus-infected cell from attack by natural killer cells. BMC Immunol 2003; 4: 4.
Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC et al. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med 2003; 197: 1427–1439.
Eagle RA, Traherne JA, Hair JR, Jafferji I, Trowsdale J . ULBP6/RAET1L is an additional human NKG2D ligand. Eur J Immunol 2009; 39: 3207–3216.
Wu J, Chalupny NJ, Manley TJ, Riddell SR, Cosman D, Spies T . Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J Immunol 2003; 170: 4196–4200.
Spreu J, Stehle T, Steinle A . Human cytomegalovirus-encoded UL16 discriminates MIC molecules by their alpha2 domains. J Immunol 2006; 177: 3143–3149.
Muller S, Zocher G, Steinle A, Stehle T . Structure of the HCMV UL16-MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PLoS Pathog 2010; 6: e1000723.
Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR . NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 2009; 83: 12345–12354.
Chalupny NJ, Rein-Weston A, Dosch S, Cosman D . Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem Biophys Res Commun 2006; 346: 175–181.
Bennett NJ, Ashiru O, Morgan FJ, Pang Y, Okecha G, Eagle RA et al. Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J Immunol 2010; 185: 1093–1102.
Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M et al. Host immune system gene targeting by a viral miRNA. Science 2007; 317: 376–381.
Beck S, Barrell BG . Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature 1988; 331: 269–272.
Browne H, Churcher M, Minson T . Construction and characterization of a human cytomegalovirus mutant with the UL18 (class I homolog) gene deleted. J Virol 1992; 66: 6784–6787.
Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ . The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity 1995; 3: 583–590.
Chapman TL, Heikeman AP, Bjorkman PJ . The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity 1999; 11: 603–613.
Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH et al. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med 1998; 187: 1681–1687.
Vales-Gomez M, Shiroishi M, Maenaka K, Reyburn HT . Genetic variability of the major histocompatibility complex class I homologue encoded by human cytomegalovirus leads to differential binding to the inhibitory receptor ILT2. J Virol 2005; 79: 2251–2260.
Cerboni C, Achour A, Wärnmark A, Mousavi-Jazi M, Sandalova T, Hsu ML et al. Spontaneous mutations in the human CMV HLA class I homologue UL18 affect its binding to the inhibitory receptor LIR-1/ILT2/CD85j. Eur J Immunol 2006; 36: 732–741.
Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P et al. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J Immunol 2005; 175: 7457–7465.
Wyrwicz LS, Rychlewski L . Cytomegalovirus immediate early gene UL37 encodes a novel MHC-like protein. Acta Biochim Polon 2008; 55: 67–73.
Revilleza MJ, Wang R, Mans J, Hong M, Natarajan K, Margulies DH . How the virus outsmarts the host: function and structure of cytomegalovirus MHC-I-like molecules in the evasion of natural killer cell surveillance. J Biomed Biotechnol 2011; 2011: 724607.
Yang Z, Bjorkman PJ . Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci USA 2008; 105: 10095–10100.
Park B, Oh H, Lee S, Song Y, Shin J, Sung YC et al. The MHC class I homolog of human cytomegalovirus is resistant to down-regulation mediated by the unique short region protein (US)2, US3, US6, and US11 gene products. J Immunol 2002; 168: 3464–3469.
Samaridis J, Colonna M . Cloning of novel immunoglobulin superfamily receptors expressed on human myeloid and lymphoid cells: structural evidence for new stimulatory and inhibitory pathways. Eur J Immunol 1997; 27: 660–665.
Reyburn HT, Mandelboim O, Valés-Gómez M, Davis DM, Pazmany L, Strominger JL . The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature 1997; 386: 514–517.
Prod'homme V, Griffin C, Aicheler RJ, Wang EC, McSharry BP, Rickards CR et al. The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1− NK cells. J Immunol 2007; 178: 4473–4481.
Kaiser BK, Pizarro JC, Kerns J, Strong RK . Structural basis for NKG2A/CD94 recognition of HLA-E. Proc Natl Acad Sci USA 2008; 105: 6696–6701.
Wagner CS, Walther-Jallow L, Buentke E, Ljunggren HG, Achour A, Chambers BJ . Human cytomegalovirus-derived protein UL18 alters the phenotype and function of monocyte-derived dendritic cells. J Leuk Biol 2008; 83: 56–63.
Wagner CS, Riise GC, Bergström T, Kärre K, Carbone E, Berg L . Increased expression of leukocyte Ig-like receptor-1 and activating role of UL18 in the response to cytomegalovirus infection. J Immunol 2007; 178: 3536–3543.
Maffei M, Ghiotto F, Occhino M, Bono M, de Santanna A, Battini L et al. Human cytomegalovirus regulates surface expression of the viral protein UL18 by means of two motifs present in the cytoplasmic tail. J Immunol 2008; 180: 969–979.
Corrales-Aguilar E, Trilling M, Hunold K, Fiedler M, Le VT, Reinhard H et al. Human cytomegalovirus Fcgamma binding proteins gp34 and gp68 antagonize Fcgamma receptors I, II and III. PLoS Pathog 2014; 10: e1004131.
Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ et al. Quantitative temporal viromics: an approach to investigate host–pathogen interaction. Cell 2014; 157: 1460–1472.
Benz C, Hengel H . MHC class I-subversive gene functions of cytomegalovirus and their regulation by interferons—an intricate balance. Virus Genes 2000; 21: 39–47.
Sinzger C, Grefte A, Plachter B, Gouw AS, The TH, Jahn G . Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J Gen Virol 1995; 76( Pt 4): 741–750.
Magri G, Muntasell A, Romo N, Sáez-Borderías A, Pende D, Geraghty DE et al. NKp46 and DNAM-1 NK-cell receptors drive the response to human cytomegalovirus-infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood 2011; 117: 848–856.
Bayer C, Varani S, Wang L, Walther P, Zhou S, Straschewski S et al. Human cytomegalovirus infection of M1 and M2 macrophages triggers inflammation and autologous T-cell proliferation. J Virol 2013; 87: 67–79.
Hengel H, Reusch U, Geginat G, Holtappels R, Ruppert T, Hellebrand E et al. Macrophages escape inhibition of major histocompatibility complex class I-dependent antigen presentation by cytomegalovirus. J Virol 2000; 74: 7861–7868.
Brennan RM, Petersen J, Neller MA, Miles JJ, Burrows JM, Smith C et al. The impact of a large and frequent deletion in the human TCR beta locus on antiviral immunity. J Immunol 2012; 188: 2742–2748.
Rajagopalan S, Long EO . A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med 1999; 189: 1093–1100.
Raftery MJ, Hitzler M, Winau F, Giese T, Plachter B, Kaufmann SH et al. Inhibition of CD1 antigen presentation by human cytomegalovirus. J Virol 2008; 82: 4308–4319.
Groh V, Steinle A, Bauer S, Spies T . Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 1998; 279: 1737–1740.
Kong Y, Cao W, Xi X, Ma C, Cui L, He W . The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood 2009; 114: 310–317.
Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ . Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature 1994; 372: 336–343.
Drakesmith H, Townsend A . The structure and function of HFE. BioEssays 2000; 22: 595–598.
Birkinshaw RW, Kjer-Nielsen L, Eckle SB, McCluskey J, Rossjohn J . MAITs, MR1 and vitamin B metabolites. Curr Opin Immunol 2014; 26: 7–13.
Oganesyan V, Oganesyan N, Terzyan S, Qu D, Dauter Z, Esmon NL et al. The crystal structure of the endothelial protein C receptor and a bound phospholipid. J Biol Chem 2002; 277: 24851–24854.
Willcox CR, Pitard V, Netzer S, Couzi L, Salim M, Silberzahn T et al. Cytomegalovirus and tumor stress surveillance by binding of a human gammadelta T cell antigen receptor to endothelial protein C receptor. Nat Immunol 2012; 13: 872–879.
Sanchez LM, Chirino AJ, Bjorkman P . Crystal structure of human ZAG, a fat-depleting factor related to MHC molecules. Science 1999; 283, 1914–1919.
Acknowledgements
Our work is supported by grants of the Deutsche Forschungsgemeinschaft (DFG He2526/7-2) and by VISTRIE (Helmholtz VH-VI-424-2).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors have no conflict of interest to disclose.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Halenius, A., Gerke, C. & Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets—but how many arrows in the quiver?. Cell Mol Immunol 12, 139–153 (2015). https://doi.org/10.1038/cmi.2014.105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2014.105
Keywords
This article is cited by
-
Benchmark of tools for in silico prediction of MHC class I and class II genotypes from NGS data
BMC Genomics (2023)
-
Natural killer cells in antiviral immunity
Nature Reviews Immunology (2022)
-
Viral immune evasins impact antigen presentation by allele-specific trapping of MHC I at the peptide-loading complex
Scientific Reports (2022)
-
Proteomic analysis of the host–pathogen interface in experimental cholera
Nature Chemical Biology (2021)
-
A slowly cleaved viral signal peptide acts as a protein-integral immune evasion domain
Nature Communications (2021)
