
Abstract
Transdifferentiation of quiescent dermal fibroblasts to secretory myofibroblasts has a central role in wound healing and pathological scar formation. This myofibroblast transdifferentiation process involves TGFβ-induced de novo synthesis of alpha smooth muscle cell actin (αSMA)+ fibers that enhance contractility as well as increased expression of extracellular matrix (ECM) proteins, including collagen and fibronectin. These processes are mediated upstream by the reactive oxygen species (ROS)-producing enzyme Nox4, whose induction by TGFβ is incompletely understood. In this study, we demonstrate that Nox4 is involved in αSMA+ fiber formation and collagen production in primary human dermal fibroblasts (hDFs) using a small-molecule inhibitor and siRNA-mediated silencing. Furthermore, TGFβ-induced signaling via Smad3 is required for myofibroblast transformation and Nox4 upregulation. Immunoprecipitation-selected reaction monitoring (IP-SRM) assays of the activated Smad3 complex suggest that it couples with the epigenetic reader and transcription co-activator bromodomain and extraterminal (BET) domain containing protein 4 (BRD4) to promote Nox4 transcription. In addition, cyclin-dependent kinase 9 (CDK9), a component of positive transcription elongation factor, binds to BRD4 after TGFβ stimulation and is also required for RNA polymerase II phosphorylation and Nox4 transcription regulation. Surprisingly, BRD4 depletion decreases myofibroblast differentiation but does not affect collagen or fibronectin expression in primary skin fibroblasts, whereas knockdown of CDK9 decreases all myofibroblast genes. We observe enhanced numbers and persistence of myofibroblast formation and TGFβ signaling in hypertrophic scars. BRD4 inhibition reverses hypertrophic skin fibroblast transdifferentiation to myofibroblasts. Our data indicate that BRD4 and CDK9 have independent, coordinated roles in promoting the myofibroblast transition and suggest that inhibition of the Smad3-BRD4 pathway may be a useful strategy to limit hypertrophic scar formation after burn injury.
Similar content being viewed by others


Main
Cutaneous wound healing is a multistep process involving sequential phases of coagulation/inflammation, proliferation/re-epithelization and wound closure/remodeling.1 The proliferation phase is initiated by formation of granulation tissue providing a matrix for recruitment of activated myofibroblasts that have central role in matrix deposition, re-epithelialization and eventual wound closure.2 In certain injuries, notably burn injuries, an exaggerated response results in excessive extracellular matrix (ECM) deposition and hypertrophic scar (HTS) formation.3 HTS produces significant morbidity through pruritis, compression, anatomic deformity and decreased joint mobility for which few effective treatments are available.4 Consequently, there is a need to understand molecular mechanisms involved in HTS formation to modify the repair process and prevent these adverse outcomes.
Myofibroblasts, present in limited numbers in the normal skin, are found in overabundance in HTS after burn injury.5 Myofibroblasts express αSMA and actin-associated SM22α, providing contractile strength6, 7 as well as collagen (Col)-1 and -3 and fibronectin for ECM remodeling.8 TGFβ is a potent cytokine upregulated after injury that promotes myofibroblast transdifferentiation.9 TGFβ1 binds to the TGFβRII receptor (TβRII) which heterodimerizes with TβRI/ALK5, a complex that recruits and phosphorylates transcription factors Smad2 and Smad3 on serine residues.10, 11 Phospho-Smad2/3/4 complexes translocate into the nucleus to bind Smad-binding elements (SBEs) in gene regulatory elements to promote myofibroblast programming.12, 13, 14
In pulmonary and cardiac fibroblasts, TGFβ induces generation of reactive oxygen species (ROS) that mediate myofibroblast transdifferentiation.15, 16 TGFβ upregulates NADPH oxidase (Nox) 4 that utilizes electrons from NADPH to generate superoxide before rapidly converting it into hydrogen peroxide.17 Unlike other members of Nox family of enzymes, Nox4 is regulated at the level of gene expression.18 Suppression of Nox4 decreases myofibroblast formation and fibrosis in lung, liver, kidney and cardiac injury models.15, 19, 20, 21 However, fibroblasts are phenotypically and functionally distinct in different organ types.22 In this study, we investigated whether dermal fibroblasts also utilize Nox4 to promote myofibroblast transdifferentiation program.
Myofibroblast transdifferentiation occurs within the context of an inflammatory response. Our group has demonstrated the central role of regulated transcription elongation in mediating inducible genes in inflammatory and growth factor pathways.23, 24, 25 In this process, signal transduction pathways activate a complex of cyclin-dependent kinase 9 (CDK9) and bromodomain-containing protein 4 (BRD4) to promote active transcriptional elongation.26 Here we report the finding that Nox4 is required for TGFβ-induced transdifferentiation of human dermal fibroblasts (hDFs) into myofibroblasts via Smad3-dependent recruitment of BRD4 and CDK9. Both BRD4 and CDK9 were required for myofibroblast phenotype, whereas only CDK9 was required for ECM expression. Finally, we demonstrate that HTS fibroblasts have constitutive Smad3-BRD4 activation and inhibition of BRD4 reverses the myofibroblast phenotype. These data suggest a cooperative, independent roles of CDK9 and BRD4 in the HTS myofibroblast phenotype.
Results
TGFβ promotes dermal fibroblast Nox4 expression and myofibroblast transdifferentiation
To determine the extent of myofibroblast population in non-burn skin (NBS) and in HTS, we performed immunofluorescence staining for the myofibroblast marker αSMA27 on skin biopsies of patients taken 12–48 months following burn injury (Figure 1a). Few myofibroblasts were present in the NBS biopsies (Figure 1a). By contrast, myofibroblasts were highly abundant in the deep dermal layer of HTS at 12–24 months but disappear by 48 months after injury. We also observed robust TGFβ staining in the deep dermis at 12–24 months (Figure 1b), suggesting that the myofibroblasts are maintained in a TGFβ-rich microenvironment.

TGFβ induces dermal fibroblast Nox4 expression and promotes transdifferentiation to myofibroblast. (a) Immunofluorescence detection of αSMA+ myofibroblasts in skin biopsies taken from burn patients, 12–48 months after injury, at the site of HTS formation and from the adjacent NBS. Positive staining for αSMA is in red and counterstaining for nuclei is in blue. Note that myofibroblasts have almost completely disappeared from the dermis 48 months after injury. There is some positive staining of glandular smooth muscle cells and vascular smooth muscle cells with anti-αSMA antibody in NBS and HTS sections, which serves as an internal positive control. Scale bar represents 100 microns. (b) IHC for TGFβ in HTS and NBS tissue sections from burn patients. Positive staining for TGFβ is brown and counterstaining for nuclei is blue. Most of the positive staining for TGFβ was observed in HTS biopsies at 12–24 months in the same location as the highest amount of αSMA+ staining. Scale bar represents 100 microns. IHC was performed on up to three tissue sections from a patient at each time point. Representative images are shown. (c) hDFs were stimulated with TGFβ (10 ng/ml) for 0–48 h, and changes in gene expression for SM22α, Nox4, fibronectin and Col1α1 were analyzed via quantitative real-time PCR. Gene expression was normalized to DNA polymerase β mRNA. Data are presented as mean±S.E.M. This experiment was repeated three times. *P<0.05 versus 0 h. (d) hDF cells were seeded on coverslips, pretreated with 10 μM ALK5i LY2157299 before being stimulated with TGFβ (10 ng/ml) for up to 48 h. Immunostaining for αSMA (green) and staining for filamentous actin using AlexaFluor-568-conjugated phalloidin (red) was performed. αSMA+ cells (green or yellow fibers) were considered to be myofibroblasts. Scale bar represents 100 microns. This experiment was repeated twice and similar results were observed. (e) hDF cells were pretreated with vehicle or 10 μM ALK5i and then stimulated with TGFβ (10 ng/ml). Whole-cell extracts were collected after 24 h and western blotting analysis was performed to analyze de novo αSMA production and phospho-Smad2/3. GAPDH and Smad2/3 served as loading controls. Experiment was repeated twice and similar results were obtained
hDFs were stimulated with TGFβ to determine the changes in Nox4 expression and the myofibroblast gene program. Sm22α increased steadily and peaked at 48 h (40-fold increase versus baseline), whereas Nox4 expression peaked earlier at 12 h (138-fold increase versus baseline), gradually declining thereafter (Figure 1c). Fibronectin and Col1α1 mRNAs also increased. To determine the role of canonical TGFβ signaling, we examined the effects of a TβRI/ALK5 inhibitor (ALK5i) on TGFβ-induced αSMA stress fiber formation and gene expression. TGFβ induced stress fibers in ~30% of the cells 24 h after stimulation and ~50% by 48 h (Figure 1d), while ALK5i completely blocked stress fiber formation (Figure 1d), phospho-Smad2/3 induction and de novo αSMA synthesis (Figure 1e). These results suggest that TGFβ induces myofibroblast transdifferentiation via ALK5-mediated phospho-Smad2/3 activation.
Nox4 inhibition blocks myofibroblast differentiation with limited effect on myofibroblast genes
To probe the role of Nox4 in myofibroblast transdifferentiation, we used a potent small-molecule inhibitor, GKT137831.28 TGFβ stimulation increased the fraction of αSMA+ myofibroblasts from 0.77 to 42.6% of total cells, which was reduced to 13.2% by GKT137831 pretreatment (Figure 2a). Surprisingly, increases in SM22α, Nox4 and fibronectin mRNAs were unaltered by GKT137831 treatment and only a slight reduction in Col1α1 was seen (Figure 2b). GKT137831 treatment prevented TGFβ-induced collagen gel contraction over 48 h (Figure 2c).

Nox4 inhibition with GKT137831 and Nox4 suppression with siRNA decrease dermal myofibroblast transformation. (a) hDFs were pretreated with vehicle or GKT137831 (20 μM) for 1 h before stimulation with TGFβ for 48 h. Immunofluoresence staining for αSMA (green) and filamentous actin stain with phalloidin (red) was performed. This experiment was repeated three times and the percentage of αSMA+ myofibroblasts were quantified over the three experiments. (b) hDF cells were pretreated with vehicle or GKT137831 (20 μM) for 1 h followed by TGFβ (10 ng/ml) for 24 h. Cellular mRNA was analyzed via quantitative real-time PCR (qRT-PCR) for changes in gene expression of SM22α, Nox4, fibronectin and Col1α1. (c) Collagen gel contraction assay was performed with hDF cells. Cells were treated with vehicle or GKT137831 (20 μM) in the presence or absence of TGFβ (10 ng/ml). Experimental groups were evaluated in triplicate. Change in gel surface area was determined after 48 h and is represented as the percentage of contraction of the gel. Experiments in panels (a−c) were repeated at least three times. Data are presented as mean±S.E.M. **P<0.05 versus −TGFβ; #P<0.05 versus vehicle+TGFβ treatment. (d) After electroporation of control or Nox4 siRNAs, hDF cells were stimulated with TGFβ (10 ng/ml) for 24 h. Total RNA was collected to analyze changes in gene expression by qRT-PCR. (e) hDF cells were electroporated with control or Nox4 siRNA and, after 3 days, were stimulated with TGFβ for 48 h. Immunofluoresence staining for αSMA (green) and phalloidin staining for filamentous actin (red) was performed to detect myofibroblasts. The percentage of cells that transdifferentiate to myofibroblasts was determined. The average of three experiments is presented in the bar graph. (f) qRT-PCR for changes in SM22α, fibronectin and Col1α1 mRNAs. (g) To detect changes in ROS, DCF-DA assay was performed on cells electroporated with control or Nox4 siRNA and incubated with or without TGFβ (10 ng/ml). As a positive control, hDF cells electroporated with control siRNA were treated with 4.8 nM H202. ROS detection assay was performed twice and each treatment group was evaluated in quadruplicates. Immunofluorescence staining in panel (d) and qRT-PCR experiments in panels (e and f) were performed three times each. All data are presented as mean±S.E.M. **P<0.05 versus −TGFβ; #P<0.05 versus control siRNA+TGFβ
To assess the specific role of Nox4, we depleted Nox4 mRNA by siRNA transfection. Nox4 expression was reduced by >80% in untreated hDFs and was significantly reduced in response to TGFβ (Figure 2d). Nox4 silencing decreased TGFβ-induced αSMA+ myofibroblast differentiation >50% versus control siRNA-treated cells (Figure 2e). Nox4 depletion did not affect TGFβ-induced changes in myofibroblast genes SM22α or fibronectin but did inhibit the induction of Col1α1. To verify that Nox4 mediated ROS generation, we directly measured ROS by dichlorofluorescein (DCF) assay.29, 30 TGFβ induced DCF fluorescence above baseline levels; this induction was completely blunted in Nox4-silenced cells without affecting basal levels (Figure 2f). This suggests that TGFβ-induced ROS is mediated through Nox4.
Smad3 regulates Nox4 and myofibroblast transformation
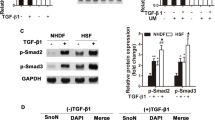
We next asked whether Smad3 was essential in Nox4 gene expression and in myofibroblast transdifferentiation. We observed a 70–80% depletion of Smad3 with siRNA (Figure 3a). Interestingly, we also noted that TGFβ decreased Smad3 mRNA by >80% after 24 h, suggesting the presence of a negative feedback loop. Smad3 silencing decreased myofibroblast formation by >75% compared with control siRNA-treated cells (Figure 3b). In addition, Smad3 silencing blocked TGFβ induction of SM22α mRNA, whereas the expression of Nox4, fibronectin and Col1α1 genes was reduced by 25–30% versus control siRNA-treated cells (Figure 3c).

Smad3 regulates myofibroblast transdifferentiation and binds to BRD4 during TGFβ stimulation. (a) hDFs were electroporated with control or Smad3 siRNA. After 72 h incubation, cells were stimulated with TGFβ (10 ng/ml) for 48 h before analysis. Top panel, whole-cell extracts from hDF were analyzed for Smad3 content by western blotting. GAPDH was used as a loading control. Bottom panel, Smad3 mRNA expression was analyzed by quantitative real-time PCR (qRT-PCR). Immunofluoresence and qRT-PCR experiments were repeated at least three times. Western blotting was repeated twice. (b) hDF cells were fixed and immunostained for αSMA (green) and phalloidin for filamentous actin (red). Myofibroblasts were quantified in three separate experiments. Data presented are the mean of three experiments. (c) qRT-PCR of myofibroblast and ECM gene expression. Data in panels (a−c) are presented as mean±S.E.M. **P<0.05 versus −TGFβ; #P<0.05 versus vehicle+TGFβ . (d) Immunofluoresence staining for Smad2/3, phospho-Smad2/3, BRD4 and CDK9 in hDF cells incubated with or without TGFβ (10 ng/ml) for 24 h. DAPI was used to stain nuclei. Scale bar represents 100 microns. Immunofluoresence experiments were repeated twice and similar results were observed. (e) hDFs were treated with TGFβ (10 ng/ml) for 24 h. Equal amount of cell lysates were immunoprecipitated with anti-Smad3 antibody and subjected to SID-SRM-MS analysis of Smad3 and BRD4 protein levels. Data are presented as the mean ratio of native to SIS peptides or as BRD4 signal normalized to Smad3. (f) hDF cells were treated with TGFβ (10 ng/ml) for 24 h and cell lysates were immunoprecipitated with an anti-BRD4 antibody before being subjected to SID-SRM-MS analysis for BRD4, Smad3 and CDK9 proteins. Data are presented as the mean ratio of native to SIS peptide. All immunoprecipitation experiments were repeated three times. Bar graphs represent mean±S.E.M. *P<0.05 versus –TGFβ
Smad3 directly interacts with the transcriptional co-activator BRD4
How TGFβ-induced ALK5-Smad3 pathway activates myofibroblast programs is not known. Knowing that BRD4 and CDK9 have central roles in inducible inflammatory gene expression23, 24, 25, 31, 32 – a hallmark of the injury response33 – and reasoning that these co-activators may have a central role in the initial stages of wound healing, we asked whether Smad2/3 localizes in the same cellular compartments. Smad2/3 accumulated within the nucleus after TGFβ stimulation as did phosphorylated Smad2/3 (Figure 3d). BRD4 and CDK9 were observed only in the nucleus, suggesting that their primary involvement may be in transcription regulation.
To determine whether Smad3 directly interacts with BRD4 and CDK9, whole-cell extracts were enriched for Smad3 by immunoprecipitation followed by quantitative measurement of BRD4 and CDK9 via selected reaction monitoring-mass spectrometry (SID-SRM-MS34, 35, 36, 37). We observed a significant enhancement of Smad3 in the Smad3 immunoprecipitates compared with control IgG, confirming target enrichment (Figure 3e, left panel). In a manner consistent with reduction of Smad3 mRNA expression in response to TGFβ treatment (Figure 3a), the abundance of Smad3 protein in TGFβ-treated immunoprecipitates was also reduced to ~25% of that of untreated cells (Figure 3e, left panel). SID-SRM-MS assays of the Smad3 immunoprecipitates showed that BRD4 associated with Smad3 in both unstimulated and stimulated cells (Figure 3e, middle panel). To account for the dramatic reduction in total Smad3 from TGFβ treatment, we normalized BRD4 signal to Smad3 abundance in each immunoprecipitate. This analysis showed the normalized fraction of Smad3 bound to BRD4 increased by more than three-fold after TGFβ treatment (Figure 3e, right panel). The reverse experiment, immunoprecipitating with anti-BRD4 antibody and quantifying bound Smad3, demonstrated a significant enrichment of BRD4 relative to control IgG (Figure 3f, left panel). In this case, the abundance of BRD4 was similar in control and TGFβ-treated samples (Figure 3f, left panel), and we observed also that TGFβ treatment increased Smad3 bound to BRD4 by ~3-fold (Figure 3f, middle panel) and increased CDK9 bound to BRD4 by ~4-fold (Figure 3f, right panel). These data suggest that TGFβ increases the fraction of active Smad3 complexed with BRD4 and that BRD4 interacts with CDK9.
Inhibition of BRD4 prevents myofibroblast transdifferentiation
To determine whether BRD4 was functionally required for Nox4 transcription and myofibroblast differentiation, we utilized JQ1, a competitive inhibitor of the BET bromodomain pocket used to bind to acetylated residues.38 JQ1 completely blocked the myofibroblast transition produced by TGFβ treatment (Figure 4a). At concentrations as low as 0.5 μM, JQ1 blocked not only TGFβ-induced increases in SM22α, Nox4, fibronectin and Col1α1 mRNA but also decreased their constitutive expression. JQ1 also blocked TGFβ-induced collagen gel contraction without significantly affecting baseline contraction (Figure 4c).

JQ1 treatment and BRD4 suppression with siRNA block myofibroblast transdifferentiation. (a) hDF cells were preincubated with 1 μM JQ1 or vehicle for 1 h before stimulation with TGFβ (10 ng/ml) for 48 h. Immunofluoresence staining for αSMA (green) and phalloidin staining for filamentous actin (red) was used to detect myofibroblasts. Myofibroblast cells (αSMA+) were quantified in three separate experiments. (b) hDF cells were preincubated with 0.5 or 1 μM JQ1 for 1 h and then stimulated with TGFβ (10 ng/ml) for 24 h. mRNA was analyzed via quantitative real-time PCR (qRT-PCR) to determine myofibroblast gene expression changes. (c) Collagen gel contraction assay was performed with hDF cells treated with vehicle or 1 μM JQ1 in the presence or absence of TGFβ (10 ng/ml). Gel surface area was measured at time 0 and 48 h and change in surface area is reported as the percentage of contraction of gel. The assay was performed in triplicate. Experiments in panels (a−c) were repeated at least three times. Data are reported as mean±S.E.M. *P<0.05 versus 0 μM JQ1−TGFβ; **P<0.05 versus −TGFβ #P<0.05 versus vehicle+TGFβ. (d) hDF cells were electroporated with control or BRD4 siRNA. After 72 h, cells were incubated with or without TGFβ (10 ng/ml) for another 48 h. Knockdown efficiency of BRD4 was determined via western blotting and qRT-PCR. β-Actin was used as a loading control. Western blotting analysis was repeated twice. (e) αSMA immunostaining (green) and phalloidin staining for filamentous actin (red) was performed to quantify myofibroblasts (aSMA+ cells). Myofibroblast cells were quantified in four separate experiments. (f) qRT-PCR analysis for myofibroblast genes. Data are presented as mean±S.E.M. *P<0.05 versus control siRNA−TGFβ; **P<0.05 versus −TGFβ; #P<0.05 versus control siRNA+TGFβ
Using siRNA-mediated transfection, we achieved elimination of both the long and short BRD4 variants in western blotting and at least a 80% reduction in BRD4 mRNA expression (Figure 4d). BRD4 silencing prevented αSMA+ myofibroblast differentiation (Figure 4e) and diminished TGFβ induction of SM22α and Nox4 by >50% (Figure 4f). Surprisingly, TGFβ-induced transcription of fibronectin and Col1α1 was not affected by BRD4 knockdown, suggesting that another BET family member, in addition to BRD4, is involved in regulating these myofibroblast genes.
Inhibition of CDK9 diminishes myofibroblast transdifferentiation
Because BRD4 complexes with CDK9, we next examined the role of CDK9 in myofibroblast differentiation using a CDK9-specific, small molecule inhibitor – Can508.39 Can508 successfully reduced TGFβ-mediated αSMA+ myofibroblast transformation by 80% (Figure 5a). However, Can508 inhibited baseline Nox4 mRNA transcription by >90% but had minimal effect on Nox4 mRNA in response to TGFβ stimulation (~15% reduction versus vehicle-treated cells, Figure 5b). In contrast, Can508 was a very potent inhibitor of SM22α, fibronectin and Col1α1 mRNA both at baseline and in response to TGFβ stimulation, reducing gene expression by 40–90% versus vehicle-treated cells (Figure 5b). Further, Can508 treatment reduced contraction under basal conditions and during TGFβ stimulation. These data suggest that CDK9 activity is functionally required for stress fiber formation, contractility and ECM synthesis.

Inhibition of CDK9 with Can508 or CDK9 knockdown with siRNA decreases myofibroblast transformation. (a) hDFs were pretreated with Can508 (30 μM) for 6 h before stimulation with TGFβ (10 ng/ml) for 48 h. Immunofluorescence for αSMA (green) and phalloidin staining for f-actin (red) was performed. αSMA+ myofibroblast cells were quantified in three separate experiments. (b) Myofibroblast gene expression changes were analyzed via quantitative real-time PCR (qRT-PCR) in hDF cells pretreated with Can508 and stimulated with TGFβ (10 ng/ml) for 24 h. (c) Collagen contraction assay was performed on hDF cells treated with vehicle or Can508 (30 μM) in the presence or absence of TGFβ (10 ng/ml). Change in surface area is reported as the percentage of contraction of the gels. Experiments in panels (a–c) were repeated at least three times. Data are represented as mean±S.E.M. *P<0.05 versus vehicle−TGFβ; **P<0.05 versus −TGFβ; #P<0.05 versus vehicle+TGFβ. (d) hDF cells were electroporated with control or CDK9 siRNA before being stimulated with TGFβ (10 ng/ml) for 48 h. Top panel, whole-cell extracts extracts were assayed for CDK9 expression by western blotting. GAPDH was used as a loading control. Western blotting was repeated twice with similar results being observed. Bottom panel, qRT-PCR analysis of CDK9 mRNA. Data are presented as mean±S.E.M. of at least three experiments. (e) Cells were immunostained for αSMA and also stained for f-actin with phalloidin (red) to determine the myofibroblast population. Average of five experiments is represented in the bar graph. (f) mRNA was assayed for gene expression by qRT-PCR. Data are presented as mean±S.E.M. of at least three experiments. *P<0.05 versus control siRNA−TGFβ; **P<0.05 versus −TGFβ; #P<0.05 versus control siRNA+TGFβ
As Can508 may have off-target effects, CDK9 was depleted in hDF cells to verify its role in myofibroblast differentiation. Transfection with CDK9 siRNA decreased CDK9 protein levels by >80% compared with control siRNA-transfected cells (Figure 5d); CDK9 mRNA expression was suppressed to a similar extent (Figure 5d). We found that CDK9 depletion inhibited TGFβ-induced αSMA+ myofibroblast transformation by ~50% (Figure 5e). Furthermore, CDK9 knockdown diminished TGFβ induction of Nox4, SM22α, fibronectin and Col1α1 by ~25–90% versus levels observed in control cells (Figure 5f). This demonstrated to us that CDK9 has a vital role in the transdifferentiation process by regulating myofibroblast gene transcription.
TGFβ-induced recruitment of p-Smad3 and CDK9 to the Nox4 promoter is dependent on BRD4
As TGFβ-induced transcription of Nox4 requires the transcription regulatory proteins Smad3, BRD4 and CDK9, we inferred that they must be recruited to the Nox4 proximal promoter. We examined the binding of these proteins to the Nox4 gene and their dependence on BRD4 acetylated histone reader using a two-step crosslink chromatin immunoprecipitation (XChIP) assay.40 TGFβ treatment led to a 4.3-fold enrichment of activated p-Smad3 on Nox4 promoter (Figure 6a). Interestingly, inducible Smad3 recruitment was blocked by JQ1. Concordantly, TGFβ induced a striking 12-fold increase in BRD4 binding (Figure 6b). JQ1 reduced BRD4 accumulation to 1.9-fold, indicating that BRD4 bromodomain interactions mediated its recruitment. TGFβ also induced a 6.4-fold enrichment of CDK9 binding to Nox4 relative to untreated control hDFs, an effect that was also blocked by JQ1 (Figure 6c). Because BRD4 and CDK9 are both capable of phosphorylating Pol II at Ser2 to promote transcription elongation, we assessed the effect of TGFβ on p-Ser2-Pol II enrichment. TGFβ induced a 2.6-fold enrichment of p-Ser2-Pol II on the Nox4 gene; JQ1 treatment completely blocked this enrichment (Figure 6d). These data indicate that TGFβ-induced recruitment of Smad3–BRD4–CDK9 complex is BRD4 dependent and that this complex is a key regulatory step in myofibroblast transdifferentiation promoting p-Ser2-Pol II formation and transcription elongation of Nox4 mRNA expression.

Increased accumulation of Smad3, BRD4 and CDK9 on Nox4 promoter after TGFβ stimulation. hDFs were preincubated with vehicle or JQ1 for 1 h before being stimulated with TGFβ (10 ng/ml) for 24 h. Cells were then subjected to XChIP analysis. (a) pSmad3 binding on the Nox4 promoter. Chromatin was immunoprecipitated with an antibody to p-Smad3; non-specific IgG was used as a negative control. Fold change was determined compared with −TGFβ sample. (b) BRD4 binding. Experiment as in panel (a). (c) CK9 binding. (d) phospho-Ser2 RNA Pol II. For experiments in panels (a–d), data are presented as mean±S.E.M. from two independent experiments; similar results were obtained in each
Increased sensitivity of HTS fibroblasts to TGFβ is mediated by BRD4
We have previously demonstrated that HTS fibroblasts are hypersensitive to IL-6 trans-signaling and express higher levels of ECM genes and cell proliferation markers,41 indicating that HTS fibroblasts are reprogrammed to express the fibrotic phenotype. Therefore, we asked whether HTS fibroblasts also display an exaggerated response to TGFβ and whether this phenotypic behavior is BRD4 dependent. We observed a greater fraction of myofibroblasts in HTS cultures than NBS under basal conditions (Figure 7a). Importantly, JQ1 treatment reverted most of the myofibroblast population back to quiescent fibroblast state (Figure 7a). Strikingly, incubation with TGFβ increased the myofibroblast population 4–5-fold in both cell types but co-treatment with JQ1 not only blocked the induction of αSMA but also decreased the myofibroblast population to levels observed with JQ1 treatment alone. Nox4, fibronectin and Col1α1 expression was significantly elevated (2.5–3-fold) at baseline in HTS fibroblasts versus NBS fibroblasts (Figure 7b). Furthermore, TGFβ induced elevation in mRNA of all four genes but HTS fibroblast displayed an even higher induction (by ~2-fold) of ECM genes fibronectin and Col1α1. JQ1 co-treatment blocked TGFβ induction of myofibroblast genes or even suppressed them below baseline levels. Because we identified SM22α and Nox4 gene transcription to be BRD4 dependent in hDFs, we asked whether BRD4 also controlled their transcription in HTS fibroblasts. BRD4 silencing suppressed TGFβ induction of both SM22α and Nox4 by >50% (Figure 7c). Collectively, these data demonstrate that HTS fibroblasts have elevated expression of myofibroblast genes in the presence and absence of TGFβ and that these cells functionally require BRD4 to maintain their state of transdifferentiation.

HTS fibroblasts have increased propensity for myofibroblast transformation, which can be blocked with JQ1. (a) HTS and NBS fibroblasts were preincubated with vehicle or JQ1 (1 μM) for 1 h before being stimulated with TGFβ (10 ng/ml) for 48 h. Cells were then immunostained for αSMA (green) and co-stained for f-actin with phalloidin (red). αSMA+ myofiboblasts were quantified in three separate experiments. Data are presented as mean±S.E.M. *P=0.055 versus NBSF; **P<0.05 versus unstimulated cells; #P<0.05 versus TGFβ. (b) NBS and HTS fibroblasts were subjected to quantitative real-time PCR (qRT-PCR) analysis of myofibroblast gene expression changes after incubation with or without JQ1 (1 μM) and TGFβ (10 ng/ml). The experiment was repeated three times. Data are presented as mean±S.E.M. *P<0.05 versus NBSF; **P<0.05 versus unstimulated cells; #P<0.05 versus TGFβ. (c) BRD4 knockdown with siRNA was performed in HTS fibroblasts before TGFβ (10 ng/ml) stimulation for 24 h. Cellular mRNA was extracted and qRT-PCR was performed for BRD4-dependent myofibroblast genes. This experiment was repeated three times. Data are presented as mean±S.E.M. **P<0.05 versus control siRNA−TGFβ; #P<0.05 versus control siRNA+TGFβ. (d) XChIP analysis for Smad3 and BRD4 on the Nox4 promoter was performed on NBS and HTS fibroblasts incubated with or without TGFβ (10 ng/ml) for 6 h. Fold change is calculated relative to NBSF−TGFβ sample. XChIP experiment was performed twice and similar results were observed. Data are presented as mean±S.E.M. (e) XChIP for CDK9 binding to the Nox4 promoter. Experiment is carried out as in panel (d)
HTS fibroblasts have increased Smad3 and BRD4 binding on the Nox4 promoter
Using XChIP analysis, we observed an ~3-fold increase of Smad3 binding to the Nox4 promoter in HTS cells (Figure 7d), and TGFβ-induced enrichment of Smad3 was enhanced HTS cells (5.2- versus 2.7-fold in NBS fibroblasts). Similarly, there was more constitutive BRD4 binding to the Nox4 gene in HTS fibroblasts – a 2.3-fold increase compared with NBS fibroblasts. TGFβ also induced enhanced BRD4 on the Nox4 gene in HTS versus NBS fibroblasts (5.8- versus 2.8-fold, respectively). Similar to the BRD4-binding pattern, there was also more constitutive and TGFβ-induced CDK9 binding to Nox 4 in HTS fibroblasts versus NBS fibroblasts (Figure 7e), indicating hyper-responsiveness of the transcriptional elongation complex in HTS. These data suggest that elevated expression of myofibroblast genes in HTS fibroblasts is due to increased basal accumulation of the Smad3–BRD4 complex promoting enhanced transcription.
Discussion
HTS is a devastating sequela of burn injury characterized by an overabundance of αSMA+ myofibroblasts and excess deposition of ECM. In this study, we verified that persistence of myofibroblasts in HTS dermis is driven by enhanced TGFβ expression in its microenvironment. Dermal fibroblasts develop αSMA+ stress fibers, highly upregulate Nox4 and SM22α and increase transcription of fibronectin and Col1α1 after exposure to TGFβ. We demonstrate that Nox4 is involved in αSMA+ stress fiber formation and collagen transcription in hDF cells. Nox4 transcription is not only controlled by Smad3 during TGFβ stimulation but also requires association with the BRD4 epigenetic reader and transcription co-activator. BRD4 interacts with both Smad3 and CDK9, part of P-TEFb complex necessary for phosphorylation on the Ser2 residue of the heptad repeat to induce full-length Nox4 transcripts. We found that HTS fibroblasts express high levels of myofibroblast genes, including Nox4, under basal conditions. HTS have a more robust response of the ECM genes to TGFβ than NBS fibroblasts – changes that can be inhibited by JQ1. Mechanistically, HTS fibroblasts have more Smad3, BRD4 and CDK9 binding to the Nox4 promoter even in the absence of TGFβ, suggesting that overactivation of the canonical Smad pathway may be the reason for the hyperresponsive cellular phenotype.
Either inhibition of Nox4 activity with GKT13831 or its suppression with siRNA had limited effect on myofibroblast genes in hDF cells. Nox4 was necessary for induction of αSMA and collagen but dispensable for SM22α and fibronectin. Previous work using fetal and adult lung fibroblasts15 and cardiac fibroblasts16 suggested a role of Nox4-generated ROS in the regulation of fibronectin. The discrepancy may reflect phenotypic differences of the fibroblast based on their origin. A global gene expression analysis of human fibroblasts from 43 different anatomical sites suggests systematic differences in the gene expression with respect to anterior–posterior, proximal–distal and dermal–nondermal origins.22 Similar to our observations in HTS fibroblasts, in dermal fibroblasts from systemic sclerosis patients, Nox4 was highly expressed and was upstream of Col1 expression.42 Surprisingly, Nox4 deficiency in mice delayed wound closure but did not affect the presence of αSMA+ myofibroblasts over a 2 week period, suggesting that αSMA induction is not entirely Nox4 dependent.43
Although TGFβ-induced Nox4 expression is primarily controlled by a SBE 4 kb upstream of the transcriptional start site of the gene,44 our findings that Smad3 inhibition does not completely block Nox4 expression in hDF cells suggests the role of additional regulatory factors. Recent work has identified contributory roles of the CArG box, the binding element of the myocardin-related transcription factor,45 as well as the transcription factors TAZ/YAP in the Hippo pathway,45 and Sp346 in regulating Nox4 in a cell-type-dependent manner. More work will be required to understand the role, if any, of these transcription factors in hDF cells.
Nevertheless, because Smad3 is an important regulator of Nox4 and the myofibroblast program (Figure 3b), we investigated the role of Smad3 transcription co-activators in this study. We demonstrate for the first time that Smad3 binds to BRD4 in response to TGFβ stimulation and that BRD4 is functionally required for expression of a subset of Smad3-dependent myofibroblast genes. Although BRD4 is well established to be a component of the PTEFb transcriptional elongation complex,47, 48 our studies indicate that BRD4 and CDK9 function in myofibroblast transdifferentiation in independent manners. This conclusion is based on the observations that BRD4 tightly binds to Smad3, whereas CDK9 does not and that BRD4 is dispensable for ECM gene expression, whereas CDK9 is needed. These functional differences are in contrast to the common pattern of BRD4 and CDK9 binding to the Nox4 promoter in HTS and respond to TGFβ by further enhanced binding to the Nox4 promoter. In addition to its role as an acetylate histone adaptor promoting PTEFb binding to RNA Pol II, BRD4 also facilitates Mediator recruitment to promote transcriptional enhancement and separately is involved as a histone acetyl transferase stimulating nucleosome clearance during transcriptional elongation.49 More work will be required to dissect the contributions of these individual activities to myofibroblast gene expression.
The mechanism for Smad3–BRD4 association is not known. BRD4 binds acetylated histones and acetylated transcription factors such as TWIST and RelA/NF-κB, suggesting that acetylated histones and transcription factor serve as important signals for recruitment of transcription machinery important in mesenchymal gene expression programs.31, 50, 51, 52 We note that the p300/CBP histone acetyltransferase acetylates Lys-372 and Lys-19 of Smad3 required for Smad3 transcriptional activity. We suspect that one or both of these acetylated residues may serve as docking sites for BRD4, but more work is needed to demonstrate this point. Although JQ1 inhibited all myofibroblast gene expression, BRD4 suppression decreased αSMA fibers and Nox4 and SM22 mRNA but not fibronectin and Col1α1 mRNA. This finding suggests that other BET proteins such as BRD2 may be involved in myofibroblast transformation of hDFs.53
P-TEFb is a complex of CyclinT1 and CDK9 that phosphorylates Pol II on Ser2 to promote transcription elongation during activation of innate immunity.26 Here we demonstrate that CDK9 also is important in the expression of myofibroblast genes and specifically for Nox4 expression. TGFβ induced the enrichment of CDK9 and promoted phosphorylation of Pol II Ser2 on the Nox4 promoter. Although we were unable to detect CDK9 in Smad3 immunoprecipitates, we found that CDK9 suppression inhibited the expression of myofibroblast genes that were also Smad3 dependent. This suggests that Smad3 may not directly interact with CDK9, although CDK9 is functionally required for induction of Smad3-dependent genes.
We have previously demonstrated that HTS fibroblasts express higher levels of gp130 – part of the IL-6 receptor complex – and exhibit an exaggerated response to IL-6 leading to higher expression of Col1α2 and fibronectin.41 Here we demonstrate that HTS fibroblasts express high levels of Nox4, fibronectin and Col1α1 under basal conditions and upregulate the expression even more when exposed to TGFβ. Our data suggest that increased basal activation of the Smad pathway coupled with BRD4 on the Nox4 gene promoter drives the myofibroblast phenotype in HTS cells. Recently, it has been reported that JQ1 administration before or after onset of hepatic fibrosis in a mouse model limits ECM deposition and decreases myofibroblast markers.54 Collectively, these finding support an important role of Smad signaling and BET proteins, including BRD4, in myofibroblast transdifferentiation and fibrosis. These findings suggest that inhibitors of the Smad3–BRD4 pathway may be useful in limiting HTS after burn injury.
Materials and methods
Cell culture and tissue biopsies
Biopsies of non-burned skin and postburn HTSs were acquired from pediatric burn patients (aged 0–18 years) at 12, 24 and 48 months following burn injuries covering at least 30% of the total body surface as part of a study approved by IRB at the University of Texas Medical Branch. After obtaining patient consent, biopsies were taken from the site of HTS or the adjacent NBS region during surgical revision procedure. Samples were formalin fixed, paraffin embedded and sectioned at 4 μ thickness. Fibroblasts from HTS and NBS biopsies were isolated as previously described55 and propagated in DMEM containing 15% fetal bovine serum (FBS) and 1% antibiotic/antimycotic (Invitrogen, Carlsbad, CA, USA). Experiments were performed on NBS and HTS cells from equivalent 6–15 passages. Cells were not tested for mycoplasma contamination. For experiments involving TGFβ, NBS and HTS cells were serum starved overnight in media containing 0.5% FBS. Normal adult hDF were purchased from Lonza and cultured in the manufacturer recommended media. All experiments with hDF cells were performed on cells from passage 4 to 12.
Reagents and antibodies
Human TGFβ1 (Peprotech, no. 100-21) was suspended in vehicle containing BSA, as recommended by the manufacturer, at 10 μg/ml stock concentration. Aliquots were kept frozen at −20 °C until used. GKT137831 was a gift from Genkyotex (Geneva, Switzerland), JQ1 and LY2157299 were purchased from Apexbio (Houston, TX, USA) and Can508 was purchased from Santa Cruz (Dallas, TX, USA).
The following primary antibodies were used for immunohistochemistry (IHC) or immunofluorescence: anti-TGFβ (Abcam, ab66043, 1:100), anti-SMA (Abcam, Cambridge, MA, USA, ab5694, 1:100), anti-Smad2/3 (Cell Signaling, no. 8685, 1:300), anti-phospho-Smad2/3 (Cell Signaling, Danvers, MA, USA,no. 8828, 1:200), anti-BRD4 (Millipore, Tenecula, CA, USA, ABE1391, 1:100), and anti-CDK9 (Santa Cruz, sc-484, 1:100). Fluorescent secondary antibodies utilized were highly cross-adsorbed goat anti-rabbit IgG or goat anti-mouse IgG conjugated to AlexaFluor (AF)-568 or AF-488 (Molecular Probes, Eugene, OR). Cell Signaling antibodies (Anti-Smad2/3 and anti-phospho-Smad2/3) and Santa Cruz antibody (anti-CDK9) were also used for western blottings. Other antibodies used in western blotting analyses were raised against BRD4 (Invitrogen, 23476, 1:1000), GAPDH (Millipore, MAB374, 1:1000) and β-actin (Sigma St. Louis, MO, USA, A5316, 1 : 5000) or were HRP-conjugated secondary antibodies (GE Healthcare, Pittsburgh, PA, USA) raised against rabbit or mouse IgGs. For XChIP analysis, anti-P-Smad3 (Cell Signaling, no. 9520), anti-BRD4 (Millipore), anti-CDK9 (Santa Cruz) and anti-Phospho-Pol II (Abcam, ab5095) antibodies were used.
Quantitative RT-PCR
hDF and NBS/HTS fibroblasts were seeded in six-well plates at 80% confluency and serum starved overnight with media containing 1% FBS. For experiments involving inhibitors, LY2157299 (10 μM), GKT137831 (20 μM) and JQ1 (0.5-1 μM) were added to media 1 h before TGFβ while Can508 (30 μM) was added 6 h before to ensure maximum inhibition.24 Fibroblasts were stimulated with hTGFβ1 for 24–48 h. Cellular RNA was extracted from fibroblasts using Tri Reagent (Sigma) according to the manufacturer’s instructions and 1 μg RNA was reverse transcribed using Super Script III First-Strand Synthesis System (Invitrogen). The resultant cDNA was diluted 1 : 2 with RNAase free water and 1 μl cDNA was amplified in a 10 μl reaction containing 5 μl SYBR Green Super Mix and 500 nM primers. The reaction mixtures were aliquoted in triplicates into a Bio-Rad 96-well plate. Bio-Rad CFX96 Real-Time thermal cycler was used to run the real-time reactions according to the following protocol: 95 °C for 3 min, followed by 40 cycles of 10 s at 95 °C, 30 s at 55 °C, and the reaction was terminated at 95 °C for 10 s. PCR products were subjected to melting curve analysis to ensure that only a single product was formed. Changes in gene expression were determined using ΔΔCT method. DNA Polymerase β was utilized as the housekeeping gene. The human primers used in the qPCR reactions were SM22 forward: 5′-CCGTGGAGATCCCAACTGG-3′, SM22 reverse: 5′-CCATCTGAAGGCCAATGACAT-3′; Nox4 forward: 5′-CAGATGTTGGGGCTAGGATTG-3′, Nox4 reverse: 5′-GAGTGTTCGGCACATGGGTA-3′; fibronectin forward: 5′-AGGAAGCCGAGGTTTTAACTG-3′, fibronectin reverse: 5′-AGGACGCTCATAAGTGTCACC-3′; Col1α1 forward: 5′-GTGCGATGACGTGATCTGTGA-3′, Col1α1 reverse: 5′-CGGTGGTTTCTTGGTCGGT-3′; Smad3 forward: 5′-CCATCTCCTACTACGAGCTGAA-3′, Smad3 reverse: 5′-CACTGCTGCATTCCTGTTGAC-3′; BRD4 forward: 5′-ACCTCCAACCCTAACAAGCC-3′, BRD4 reverse: 5′-TTTCCATAGTGTCTTGAGCACC-3′; CDK9 forward: 5′-GGGCTGTTGAGCAATGTTTTG-3′, CDK9 reverse: 5′-GCAGGATCTTGTTTCTGTGGA-3′; and DNA polymerase β forward: 5′-CCGCAGGAGACTCTCAACG-3′, DNA polymerase β reverse: 5′-GTACTTGTGGATAGCTTGGCTC-3′.
Western blotting
Fibroblast cells were scraped into Eppendorf tubes and lysed in RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8) containing protease (Sigma, P8340) and phosphatase inhibitor cocktails (Thermo Scientific, San Jose, CA, USA). Cellular extracts were sonicated for 10 s to ensure complete cell lysis and kept on ice for 30 min. DNA was pelleted by centrifugation at 12 000 × g at 4 °C for 20 min, and the supernatant was transferred to a separate tube. Protein samples were kept frozen at −80 °C until needed for analysis.
Protein concentrations were measured using Bradford Protein assay (Protein Reagent, Bio-Rad, Hercules, CA, USA). In all, 30–50 μg protein was fractionated by 10% SDS-PAGE and transferred to PVDF membrane. After blocking with 5% milk in TBS-Tween (TBS-T) for 1 h, membranes were incubated with primary antibody overnight at 4 °C. Membranes were washed thoroughly the next day in TBS-T and incubated with HRP-conjugated secondary antibody for 1 h. ECL Western blot solution (Amersham, Piscataway, NJ, USA) was used as the chemiluminescence substrate and the exposed X-ray film was developed with a Kodak machine (Rochester, NY, USA).
IHC on tissue sections
Tissue sections of skin biopsies from HTS and NBS regions were deparaffinized and rehydrated and antigen retrieval was performed with 10 mM sodium citrate, pH 6. Sections were blocked with 5% goat serum in TBS-T for 1 h and incubated with primary antibody overnight at 4 °C. After washing three times with TBS-T, sections were incubated with a fluorescent secondary antibody (AF-568 goat anti-rabbit) for detection of αSMA or a biotinylated secondary antibody for detecting TGFβ for 1 h. For detection via immunofluorescence, tissue sections were washed again with TBS-T, incubated with DAPI (Molecular Probes) for nuclear counterstaining and glass coverslips were mounted using Dako Fluorescence Mounting Medium (Carpenteria, CA, USA). For chromogen-based detection, tissue sections were incubated with avidin–biotin complex (Vector Labs, PK6101) followed by exposure to DAB substrate (Vector Labs, Burlingame, CA, USA, SK4100) and counterstaining with hematoxylin. Sections were dehydrated with serial washes in ethanol and xylene before mounting of coverslips. Immunostained sections were evaluated using a Nikon Eclipse 80i microscope (Melville, NY, USA) and images were captured with an attached Nikon DSM1200F digital camera. ImageJ (NIH, Bethesda, MD, USA) software was used to make composite images and to add the scale bar.
Immunocytochemistry
Fibroblast cells were seeded on coverslips at 30–40% confluency, serum starved for 16 h and stimulated with TGFβ (10 ng/ml) in the presence or absence of an inhibitor. Similar to the qRT-PCR experiments, cells were preincubated with LY2157299 (10 μM), GKT137831 (20 μM), JQ1 (0.5-1 μM) or Can508 (30 μM) before TGFβ stimulation. After 24–48 h incubation, cells were fixed in 4% paraformaldehyde and permeabilized with 0.25% Triton X-100. Cells were then blocked with 5% goat serum for 1 h and incubated with primary antibody overnight at 4 °C, followed by 1 h incubation with AF-568 or AF-488 conjugated secondary antibody. For detection of filamentous actin, AF-568-conjugated phalloidin (Molecular Probes) was added to cells after secondary antibody treatment. Cells were counterstained with DAPI and mounted on to glass slides for analysis. ImageJ (NIH) software was used to make composite images and to add the scale bar.
ROS detection assay
Nox4 activity and ROS were assessed using a DCF-DA assay as described previously with some modifications.29 Briefly, control siRNA or Nox4 siRNA-treated cells were seeded at a density of 35 K cells/well in a 24-well plate and serum starved before being treated with TGFβ. Cells were washed 2 × with PBS and incubated with 10 μM DCF-DA (Molecular Probes) for 30 min in phenol-red-free media. Following incubation with the dye, cells were washed again 2 × with PBS and incubated for another hour with fresh media. Plates were read using a Tekan Infinite F200 Pro fluorescence plate reader (ex: 480 nm, em: 520 nm). Each treatment was assessed in quadruplicate and background reading was subtracted to attain the cellular DCF fluorescence value. hDF cells treated with hydrogen peroxide (4.8 nM) for 1 h were used as positive control.
Collagen gel contraction assay
Collagen gel contraction assay was performed as previously reported.56 Briefly, fibroblasts were harvested from 10-cm plates and re-suspended in DMEM containing 1% FBS. Fifty thousand fibroblasts were then seeded into collagen matrices along with GKT137831 (20 μM), JQ1 (1 μM) or Can508 (30 μM) and cast into wells of a 24-well plate. The collagen gels were released from the edges and left floating in DMEM+1% FBS±inhibitors. Experimental groups were assessed in triplicate. Gels were photographed at 0, 24 and 48 h. Calculations based on the 48 h time point are reported in this manuscript. ImageJ software (NIH) was used to calculate the change in surface area, which is reported as the percentge of gel contraction.
Co-immunoprecipitation
Fibroblasts cells were treated with TGFβ (10 ng/ml) for 24 h before being lysed with RIPA buffer containing protease inhibitor cocktail (Sigma). Equal amounts of protein were incubated with 4 μg of control IgG or an antigen-specific antibody overnight at 4 °C under constant agitation. Next day, prewashed protein-A-conjugated magnetic beads (Invitrogen) were added to each sample and samples were incubated for 2 h at 4 °C on a tube rotator. Magnetic beads were washed 3 × with RIPA buffer and once with PBS. Immune complexes attached to the magnetic beads were pelleted and kept frozen at −80 °C until used for analysis with SID-SRM-MS.
Stable isotope dilution-selected reaction monitoring-mass spectrometry
SID-SRM-MS assays of SMAD3, CDK9 and BRD4 were developed using a workflow described in previous publications.34, 35, 36 The signature peptides and SRM parameters are listed in Table 1. The peptides were chemically synthesized incorporating isotopically labeled [13C615N4] Arginine or [13C615N2] Lysine to a 99% isotopic enrichment (Thermo Scientific). The proteins immunoprecipitated with anti-SMAD3 and anti-BRD4 antibodies were captured by protein A magnetic beads (Dynal Inc., Carlsbad, CA, USA). The proteins on the beads were digested with trypsin as described previously.34, 35, 36 Briefly, beads were washed with PBS 3 × and then resuspended in 100 μl of 50 mM ammonium hydrogen carbonate (pH 7.8) and 40 μl of 0.1 μg/μl of trypsin was added. The samples were mixed and trypsinized by gentle vortexing overnight at 37 °C. After digestion, the supernatant was collected. The beads were washed with 50 μl of 50% acetonitrile (ACN) three times and the supernatant was pooled and dried. The tryptic digests were then reconstituted in 50 μl of 4% ACN-0.01% TFA. An aliquot of 5 μl of diluted stable isotope-labeled signature peptides was added to each tryptic digest. These samples were desalted with ZipTip C18. The peptides were eluted with 80% ACN and dried with Speedvac. The peptides were reconstituted in 30 μl of 5% formic acid–0.01% TFA and were directly used for LC-SRM-MS analysis without further purification or fractionation. SRM assays were performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with nanospray source (Thermo Scientific) as described previously.34, 35, 36 The online chromatography were performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA, USA). An aliquot of 10 μl of each of the tryptic digests was injected on a C18 reverse-phase nano-HPLC column (PicoFrit, 75 μm × 10 cm; tip ID 15 μm) at a flow rate of 500 nl/min with a 20-min 98% A, followed by a 15-min linear gradient from 2 to 30% mobile phase B (0.1% formic acid–90% ACN) in mobile phase A (0.1% formic acid). The TSQ Vantage was operated in high-resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da Full Width Half Maximum. All acquisition methods used the following parameters: 2100 V ion spray voltage, a 275 °C ion transferring tube temperature, a collision-activated dissociation pressure at 1.5 mTorr, and the S-lens voltage used the values in S-lens table generated during MS calibration.
All SRM data were manually inspected to ensure peak detection and accurate integration. The chromatographic retention time and the relative product ion intensities of the analyte peptides were compared with those of the stable isotope-labeled standard (SIS) peptides. The variation of the retention time between the analyte peptides and their SIS counterparts should be within 0.05 min, and the difference in the relative product ion intensities of the analyte peptides and SIS peptides were <20%. The peak areas in the extract ion chromatography of the native and SIS version of each signature peptide were integrated using Xcalibur 2.1 (Sugarland, TX, USA). The default values for noise percentage and baseline subtraction window were used. The ratio between the peak area of native and SIS version of each peptide was calculated.
siRNA knockdown of mediators of myofibroblast transdifferentiation
hDF or HTS fibroblast were grown to 90% confluency before being harvested with 0.25% Trypsin. After trypsin neutralization, 1 × 106 cells were electroporated with 100 pmol of non-specific control siRNA (ON-TARGET plus SMARTpool by Dharmacon) or gene-specific siRNA (ON-TARGET plus SMARTpool by Dharmacon (Lafayette, CO, USA) targeting Nox4, Smad3, BRD4 or CDK9) using U-023 program (Amaxa, Walkersville, MD, USA). Cells were seeded in six-well plates or on coverslips. After 48 h, cells were serum starved overnight and then treated with TGFβ (10 ng/ml) for 24–48 h. Target knockdown by at least 75% by qRT-PCR was needed for experimental inclusion.
Dual crosslink chromatin immunoprecipitation
XChIP was performed as previously described.57 Briefly, fibroblast cells (~1.2 × 106 cells per 10-cm plate) were washed 2 × with PBS; protein–protein crosslinks were made with 2 mM disuccinimidyl glutarate (Pierce, Carlsbad, CA, USA); and protein–DNA crosslinks were formed with formaldehyde. Chromatin was sheared via five rounds of sonication at setting 4 with 10 s breaks on ice between pulses (Branson Sonifier 150, Branson Ultrasonics, Danbury, CT, USA). Equal amount of sheared chromatin was immunoprecipitated overnight at 4 °C with 4 μg of control IgG or target-specific antibody (anti-Smad3, -P-Smad3, -BRD4, -CDK9 or -P-Pol II). Protein A-conjugated magnetic beads (Invitrogen) were added to capture antibody–antigen complexes. Immunoprecipitates were washed and then eluted with elution buffer (0.09 M NaHCO3, 1% SDS). Samples were de-crosslinked in 0.2 M NaCl at 65 °C for 2–3 h. DNA was isolated using phenol–chloroform extraction and ethanol precipitation and re-suspended in TE buffer. Real-time genomic PCR was performed on the isolated DNA using primers specific for the Nox4 promoter: Nox4 forward 5′-GGACATCCTGAACAGCAGCA-3′ and Nox4 reverse 5′-CTGCACCAGTCTGCTCCG-3′. Fold change of DNA in each sample was determined by first normalizing the absolute value to the input DNA reference and then calculating the fold change relative to unstimulated cells.
Statistical analysis
Experiments were repeated at least three times, and the mean±S.E.M. of n=3 experiments was plotted, unless otherwise noted. This sample size was selected based on effect sizes and experimental variability. Differences across multiple groups were analyzed by analysis of variance, followed by Newman–Keuls or Tukey’s pairwise comparison. SigmaPlot 12.5 (Systat Software Inc., San Jose, CA, USA) was used for analyzing and graphing data. P<0.05 was considered statistically significant.
References
Singer AJ, Clark RA . Cutaneous wound healing. N Engl J Med 1999; 341: 738–746.
Darby IA, Zakuan N, Billet F, Desmouliere A . The myofibroblast, a key cell in normal and pathological tissue repair. Cell Mol Life Sci 2016; 73: 1145–1157.
Finnerty CC, Jeschke MG, Branski LK, Barret JP, Dziewsulski P, Herndon DN . Hypertrophic scarring: the greatest unmet challenge following burn injury. Lancet 2016; 388: 1427–1436.
Arno AI, Gauglitz GG, Barret JP, Jeschke MG . New molecular medicine-based scar management strategies. Burns 2014; 40: 539–551.
Nedelec B, Shankowsky H, Scott PG, Ghahary A, Tredget EE . Myofibroblasts and apoptosis in human hypertrophic scars: the effect of interferon-alpha2b. Surgery 2001; 130: 798–808.
Darby I, Skalli O, Gabbiani G . Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest 1990; 63: 21–29.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA . Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002; 3: 349–363.
Bai X, He T, Liu J, Wang Y, Fan L, Tao K et al. Loureirin B inhibits fibroblast proliferation and extracellular matrix deposition in hypertrophic scar via TGF-beta/Smad pathway. Exp Dermatol 2015; 24: 355–360.
Weber CE, Li NY, Wai PY, Kuo PC . Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J Burn Care Res 2012; 33: 311–318.
Biernacka A, Dobaczewski M, Frangogiannis NG . TGF-beta signaling in fibrosis. Growth Factors 2011; 29: 196–202.
Hata A, Chen YG . TGF-beta signaling from receptors to Smads. Cold Spring Harb Perspect Biol 2016; 8: 259–285.
Heldin CH, Miyazono K, ten DP . TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997; 390: 465–471.
Hu B, Wu Z, Phan SH . Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol 2003; 29 (3 Pt 1): 397–404.
Evans RA, Tian YC, Steadman R, Phillips AO . TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res 2003; 282: 90–100.
Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009; 15: 1077–1081.
Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005; 97: 900–907.
Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD et al. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 2011; 286: 13304–13313.
Michaeloudes C, Sukkar MB, Khorasani NM, Bhavsar PK, Chung KF . TGF-beta regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2011; 300: L295–L304.
Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernandez-Rodriguez CM, Pinedo F et al. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012; 7: e45285.
Nlandu KS, Dizin E, Sossauer G, Szanto I, Martin PY, Feraille E et al. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol 2012; 23: 1967–1976.
Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X et al. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 2015; 131: 643–655.
Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY . Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet 2006; 2: e119.
Hou T, Ray S, Brasier AR . The functional role of an interleukin 6-inducible CDK9.STAT3 complex in human gamma-fibrinogen gene expression. J Biol Chem 2007; 282: 37091–37102.
Tian B, Zhao Y, Kalita M, Edeh CB, Paessler S, Casola A et al. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J Virol 2013; 87: 7075–7092.
Brasier AR . Expanding role of cyclin dependent kinases in cytokine inducible gene expression. Cell Cycle 2008; 7: 1–6.
Peterlin BM, Price DH . Controlling the elongation phase of transcription with P-TEFb. Mol Cell 2006; 23: 297–305.
Hasan W, Zhang R, Liu M, Warn JD, Smith PG . Coordinate expression of NGF and alpha-smooth muscle actin mRNA and protein in cutaneous wound tissue of developing and adult rats. Cell Tissue Res 2000; 300: 97–109.
Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012; 56: 2316–2327.
Cai H, Dikalov S, Griendling KK, Harrison DG . Detection of reactive oxygen species and nitric oxide in vascular cells and tissues: comparison of sensitivity and specificity. Methods Mol Med 2007; 139: 293–311.
Jamaluddin M, Wang S, Boldogh I, Tian B, Brasier AR . TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal 2007; 19: 1419–1433.
Brasier AR, Tian B, Jamaluddin M, Kalita MK, Garofalo RP, Lu M . RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J Virol 2011; 85: 11752–11769.
Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S et al. RelA Ser276 phosphorylation is required for activation of a subset of NF-{kappa}B-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol 2008; 28: 3623–3638.
Newton K, Dixit VM . Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 2012; 4(3). pii: a006049.
Zhao Y, Brasier AR . Applications of selected reaction monitoring (SRM)-mass spectrometry (MS) for quantitative measurement of signaling pathways. Methods 2013; 61: 313–322.
Zhao Y, Tian B, Edeh CB, Brasier AR . Quantitation of the dynamic profiles of the innate immune response using multiplex selected reaction monitoring-mass spectrometry. Mol Cell Proteomics 2013; 12: 1513–1529.
Zhao YX, Widen SG, Jamaluddin M, Tian B, Wood TG, Edeh CB et al. Quantification of activated NF-kappa B/RelA complexes using ssDNA aptamer affinity - stable isotope dilution-selected reaction monitoring-mass spectrometry. Mol Cell Proteomics 2011; 10: 6.
Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP . Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci 2003; 100: 6940–6945.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O et al. Selective inhibition of BET bromodomains. Nature 2010; 468: 1067–1073.
Krystof V, Cankar P, Frysova I, Slouka J, Kontopidis G, Dzubak P et al. 4-Arylazo-3,5-diamino-1H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J Med Chem 2006; 49: 6500–6509.
Nowak DE, Tian B, Brasier AR . Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques 2005; 39: 715–725.
Ray S, Ju X, Sun H, Finnerty CC, Herndon DN, Brasier AR . IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J Invest Dermatol 2013; 133: 1212–1220.
Spadoni T, Svegliati Baroni S, Amico D, Albani L, Moroncini G, Avvedimento EV et al. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol 2015; 67: 1611–1622.
Levigne D, Modarressi A, Krause KH, Pittet-Cuenod B . NADPH oxidase 4 deficiency leads to impaired wound repair and reduced dityrosine-crosslinking, but does not affect myofibroblast formation. Free Radic Biol Med 2016; 96: 374–384.
Bai G, Hock TD, Logsdon N, Zhou Y, Thannickal VJ . A far-upstream AP-1/Smad binding box regulates human NOX4 promoter activation by transforming growth factor-beta. Gene 2014; 540: 62–67.
Rozycki M, Bialik JF, Speight P, Dan Q, Knudsen TE, Szeto SG et al. Myocardin-related transcription factor regulates Nox4 protein expression: linking cytoskeletal organization to redox state. J Biol Chem 2016; 291: 227–243.
Katsuyama M, Hirai H, Iwata K, Ibi M, Matsuno K, Matsumoto M et al. Sp3 transcription factor is crucial for transcriptional activation of the human NOX4 gene. FEBS J 2011; 278: 964–972.
Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG et al. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci USA 2012; 109: 6927–6932.
Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N et al. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol Cell Biol 2013; 33: 2497–2507.
Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat Struct Mol Biol 2016; 23: 540–548.
Tian B, Li X, Kalita M, Widen SG, Yang J, Bhavnani SK et al. Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics 2015; 16: 529.
Tian B, Zhao Y, Sun H, Zhang Y, Yang J, Brasier AR . Role of the Bromodomain-containing 4 epigenetic reader in mediating NFkB-dependent epithelial mesenchymal transition and pulmonary fibrosis. Am J Respir Cell Mol Biol 2016; 311: L1183–L1201.
Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014; 25: 210–225.
Tang X, Peng R, Phillips JE, Deguzman J, Ren Y, Apparsundaram S et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am J Pathol 2013; 183: 470–479.
Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M et al. BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci 2015; 112: 15713–15718.
Zhang Z, Finnerty CC, He J, Herndon DN . Smad ubiquitination regulatory factor 2 expression is enhanced in hypertrophic scar fibroblasts from burned children. Burns 2012; 38: 236–246.
Ngo P, Ramalingam P, Phillips JA, Furuta GT . Collagen gel contraction assay. Methods Mol Biol 2006; 341: 103–109.
Tian B, Yang J, Brasier AR . Two-step cross-linking for analysis of protein-chromatin interactions. Methods Mol Biol 2012; 809: 105–120.
Acknowledgements
Core laboratory support was provided by UTMB Optical Imaging Facility and the Sealy Center for Molecular Sciences Selected Reaction Monitoring Facility. This work was supported by NHLBI predoctoral award 1F30HL128036 (to TI), P50 GM060338 (to DNH), SHC 71001 (to CCF), RO1 GM112936 (to CCF), UL1TR001439 (to ARB) and NIEHS ES006676 (to ARB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by B Zhivotovsky
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ijaz, T., Jamaluddin, M., Zhao, Y. et al. Coordinate activities of BRD4 and CDK9 in the transcriptional elongation complex are required for TGFβ-induced Nox4 expression and myofibroblast transdifferentiation. Cell Death Dis 8, e2606 (2017). https://doi.org/10.1038/cddis.2016.434
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.434
This article is cited by
-
Delivery of a BET protein degrader via a CEACAM6-targeted antibody–drug conjugate inhibits tumour growth in pancreatic cancer models
Nature Communications (2024)
-
BET inhibitors drive Natural Killer activation in non-small cell lung cancer via BRD4 and SMAD3
Nature Communications (2024)
-
Bellidifolin ameliorates isoprenaline-induced cardiac hypertrophy by the Nox4/ROS signalling pathway through inhibiting BRD4
Cell Death Discovery (2023)
-
Targeting cyclin-dependent kinase 9 in cancer therapy
Acta Pharmacologica Sinica (2022)
-
Acetyl-bufalin shows potent efficacy against non-small-cell lung cancer by targeting the CDK9/STAT3 signalling pathway
British Journal of Cancer (2021)
