
Abstract
Cell death is an essential regulatory mechanism for removing unneeded cells in animal development and tissue homeostasis. The c-Jun N-terminal kinase (JNK) pathway has pivotal roles in the regulation of cell death in response to various intrinsic and extrinsic stress signals. The canonical Wingless (Wg) signaling has been implicated in cell proliferation and cell fate decisions, whereas its role in cell death remains largely elusive. Here, we report that activated Bsk (the Drosophila JNK homolog) induced cell death is mediated by the canonical Wg signaling. First, loss of Wg signaling abrogates Bsk-mediated caspase-independent cell death. Second, activation of Wg signaling promotes cell death in a caspase-independent manner. Third, activation of Bsk signaling results in upregulated transcription of wingless (wg) gene. Finally, Wg pathway participates in the physiological function of Bsk signaling in development. These findings not only reveal a previously undiscovered role of Wg signaling in Bsk-mediated cell death, but also provide a novel mechanism for the interplay between the two important signaling pathways in development.
Similar content being viewed by others

Main
Cell death plays a fundamental role in animal development and tissue homeostasis by modulating numerous physiological processes, including regulation of cell number and organ size, sculpting of tissue structures, and elimination of abnormal or aged cells.1, 2 Deregulation of cell death would lead to abnormal development and various diseases, such as tumors, neurodegenerative disorders, and immunodeficiency diseases.3, 4
c-Jun N-terminal kinase (JNK) signaling is a crucial regulatory mechanism that controls stress-induced cell death. This pathway is initiated by various intrinsic and extrinsic signals including the tumor necrosis factor (TNF) family ligands, and is mediated through a mitogen-activated protein (MAP) kinase cascade.5 Upon phosphorylation, JNK translocates into the nucleus and activates the transcription factors Jun, Fos and FoxO, which will finally induce cell death.6, 7, 8, 9 JNK pathway is highly conserved in Drosophila melanogaster, in which the TNF orthlog Eiger (Egr) triggers cell death through the JNK kinase kinase dTAK1, the JNK kinase Hemipterous (Hep), and Drosophila JNK Basket (Bsk).10, 11, 12, 13, 14 Although much progress has been made for understanding the core regulatory machine of Egr-Bsk pathway, other signaling pathways that modulate Bsk-mediated cell death have not been fully elucidated.
Wnt proteins are a family of highly conserved secreted molecules that regulate cell–cell interactions through distinct intracellular signal transduction pathways, including the canonical Wnt/β-catenin pathway, the Wnt/Ca2+ pathway, and the Wnt/Frizzled (Fz)-Planar Cell Polarity (PCP) pathway.15 The canonical Wnt signaling represents one of the best investigated pathways and has crucial roles in regulating cell proliferation, cell migration, and cell fate decisions.16, 17 Deregulated Wnt signaling is frequently associated with human diseases, especially cancers.18, 19, 20 Genetic studies of the Drosophila Wnt protein Wingless (Wg) have made great contributions to our understanding of Wnt signaling.21, 22, 23 In the absence of Wg signal, the β-catenin homolog Armadillo (Arm) is phosphorylated and degraded by a multi-protein 'destruction complex' that includes the Adenomatous Polyposis Coli protein (APC), Shaggy (Sgg)/Zeste-white-3, Axin (Axn), and Casein Kinase1 (CK1). In the presence of Wg signal, the receptor dFrizzled (dFz) and co-receptor Arrow (Arr) bind to Wg, recruit and phosphorylate the scaffold protein Dishevelled (Dsh), which antagonizes the 'destruction complex' and prevents Arm from degradation. As a consequence, accumulated Arm translocates to the nucleus and stimulates target genes expression by binding to the transcription factor Pangolin (Pan).15, 24, 25
Crosstalk between different signaling pathways occurs extensively and forms a complex signaling network that enable cells to interpret multiple inputs and execute different responses in a context-dependent manner. The interplay between JNK and the non-canonical Wnt/Frizzled (Fz)-PCP pathways has been extensively studied, in which JNK signaling acts downstream of Dsh to promote PCP establishment.26, 27, 28 Yet little is known about the interaction between JNK and the canonical Wnt/Wg pathways in development. A collaboration between JNK and the canonical Wg signaling was reported to promote dorsal closure and ventral patterning during Drosophila embryogenesis, but the two pathways appear to function in parallel.29 An interaction between AP-1 and β-catenin/TCF complex was also reported to have a role in carcinogenesis, though the underlying mechanism remains elusive.30
In the present study, we have identified from a genetic screen that the canonical Wg signaling has a crucial role in Bsk-mediated cell death in Drosophila. Loss of Wg signaling specifically suppresses Egr or Hep-induced Bsk-dependent cell death in eye, wing, and thorax. Stimulation of Wg signaling phenocopies Bsk activation and induces caspase-independent cell death. Furthermore, Wg signaling is both necessary and sufficient for the expression of Bsk signaling reporter puckered (puc). Epistasis experiments indicate that Wg signaling acts downstream of Bsk to regulate cell death and other physiological functions. Finally, activated Bsk signaling upregulates wg transcription, providing a molecular mechanism for the genetic interaction between Bsk and Wg pathways. These findings not only expand our existing knowledge about the molecular basis of complex signaling network in regulating cellular activities, but also provide additional strategy and approach for cancer therapy.
Results
Loss of Wg signaling suppresses Egr-induced small eye phenotype in Drosophila
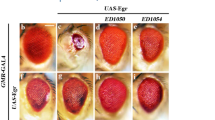
Ectopic expression of TNF ortholog Egr in the developing eye driven by GMR-Gal4 (GMR>Egr) triggers massive cell death and produces a Bsk-dependent small eye phenotype (Figures 1b, n and o),10, 11, 31 as compared with the GMR-Gal4 control (Figure 1a). To identify additional genes that modulate Egr-induced cell death, we performed a genetic screen for dominant modifiers of the GMR>Egr induced small eye phenotype.14, 32 Intriguingly, we found that this phenotype was strongly suppressed in heterozygous Sternopleural (Sp) animals (Figure 1d). Sp was originally identified as a dominant mutation that produces supernumerary sternopleural bristles,33 and was later characterized as a regulatory mutation of the wg gene.34 Consistent with this observation, the GMR>Egr induced small eye phenotype is also suppressed by two other wg mutant alleles in heterozygosity (Figure 1e and Supplementary Figure S1b) or by the expression of a wg RNAi (Figure 1i). Since wg encodes a ligand for the Wg signaling pathway, we wondered whether the canonical Wg pathway is required. Indeed, we found that the GMR>Egr induced small eye phenotype was suppressed mildly by mutation in dsh (Figure 1f and Supplementary Figure S1c), arm (Figure 1g and Supplementary Figure S1d), or pan (Figure 1h), and strongly by RNAi-mediated knocking-down of dsh (Figure 1j) or pan (Figure 1k). Furthermore, expression of Sgg or Apc2, components of the 'destruction complex' that negatively regulates Wg signaling, suppressed GMR>Egr induced small eye phenotype to a similar extent as that of BskDN (Figures 1l, m and o), a dominant-negative form of Bsk encoding the Drosophila JNK ortholog. As a negative control, GMR>Egr induced small eye phenotype is not affected by the expression of GFP (Figure 1c). Together, these results suggest that the canonical Wg pathway is required for Egr-induced small eye phenotype.

Loss of Wg signaling suppresses Egr-induced small eye phenotype. Light micrographs of Drosophila adult eyes are shown. Compared with the GMR-Gal4 control (a), GMR>Egr triggers extensive cell death and produces a small eye phenotype (b), which remains unaffected by expression of GFP (c), but is suppressed by Sp (d) or mutations in wg (e), dsh (f), arm (g), or pan (h), or RNAi knocking-down of wg (i), dsh (j), or pan (k), or overexpression of Sgg (l) or Apc2 (m). Mutation in bsk (n) and expression of BskDN (o) serve as positive controls
To examine whether Wg pathway is specifically involved in Egr-triggered cell death, we checked whether loss of Wg signaling could affect cell death triggered by Dp53, a pro-apoptotic gene encoding the Drosophila ortholog of p53.35, 36, 37 Ectopic expression of Dp53 in the eye (GMR>Dp53) induces vast cell death and produces small and rough eyes with fused ommatidia (Supplementary Figure S2a).35 However, this phenotype is not suppressed by downregulation of Wg signaling (Supplementary Figure S2), suggesting that the canonical Wg pathway is specifically required for Egr induced small eye phenotype in vivo.
Loss of Wg signaling suppresses Hep-induced Bsk-mediated small eye phenotype
Egr triggers two independent cell death pathways in Drosophila, one mediated by Bsk and another by caspases.32 To determine whether Wg pathway is required for Bsk-mediated cell death, we expressed a constitutive active form of Hep (HepCA) encoding the Drosophila JNK kinase. Expression of HepCA in the eye (GMR>HepCA) induces Bsk-dependent cell death and generates a small eye phenotype (Figure 2a).32 This phenotype is suppressed in heterozygous mutants for wg or arm (Figures 2c and d), and by knocking-down of wg, arm, or pan (Figures 2e–g) or expression of the negative regulator Sgg or Axn (Figures 2h and i), but remains unaffected by the expression of GFP (Figure 2b). As a positive control, this phenotype is suppressed in heterozygous bsk mutants (Figure 2j). Together, these data suggest that Wg signaling is required for Hep-induced small eye phenotype.

Loss of Wg signaling suppresses Hep-induced Bsk-mediated small eye phenotype. Light micrographs of Drosophila adult eyes are shown. Expression of HepCA triggers cell death and produces a small eye phenotype (a), which remains unaffected by expression of GFP (b), but is suppressed by mutations in wg (c) or arm (d), or RNAi knocking-down of wg (e), arm (f) or pan (g), or expression of Sgg (h) or Axn (i), while mutation in bsk serves as a positive control (j)
Loss of Wg signaling suppresses Bsk-mediated cell death in other tissues
To investigate whether Wg signaling modulates Bsk-mediated cell death in other tissues, we activated Bsk signaling in the dorsal thorax or wing. Expression of Hep driven by pnr-Gal4 (pnr>Hep) triggers Bsk-dependent cell death in the thorax and produces a small scutellum phenotype (Figure 3b).31 This phenotype is partially suppressed by mutation in wg, dsh, or arm (Figures 3c–e). Since knockdown of Wg signaling components in the thorax results in developmental defects (data not shown), their abilities to suppress pnr>Hep-induced scutellum phenotype could not be examined. Furthermore, expression of Hep along the anterior/posterior (A/P) compartment boundary driven by ptc-Gal4 results in elevated cell death in third instar wing discs as revealed by acridine orange (AO) staining (Figure 3l), and produces loss of the anterior cross vein (ACV) phenotype (Figure 3g). This phenotype is suppressed partially by a mutation in dsh, and near fully by the expression of a dsh RNAi, but not that of LacZ (Figures 3h–j and p). Consistently, Hep or Egr induced cell death was significantly suppressed by mutation or RNAi-mediated depletion of Wg pathway components (Figures 3m–o and q and Supplementary Figure S3). Collectively, these data suggest that the canonical Wg pathway modulates Bsk-mediated cell death in a non-tissue specific manner.

Loss of Wg signaling suppresses Bsk-mediated cell death in other tissues. Light micrographs of Drosophila adult notum (a–e) and wings (f–j), fluorescent micrographs of third instar wing discs (k–o) are shown. Compared with the pnr-Gal4 control (a), ectopic expression of Hep produces a small scutellum phenotype (b), which is partially suppressed by mutations in wg (c), dsh (d), or arm (e). Compared with the ptc-Gal4 control (f), expression of Hep produces a loss-of-ACV phenotype (g), which remains unaffected by expression of LacZ (h), but is suppressed partially by mutation in dsh (i) and strongly by RNAi knocking-down of dsh (j). Compared with the ptc-Gal4 control (k), expression of Hep induced extensive cell death (l), which remains unaffected by expression of LacZ (m), but is suppressed significantly by mutation in dsh (n) and RNAi knocking-down of dsh (o). (p) Statistics of the loss-of-ACV phenotype in (f–j). (f, 0.00%, n=203; g, 77.23%, n=101; h, 86.05%, n=86; i, 43.40%, n=53; j, 6.08%, n=148). Three asterisks, P<0.001; n.s., P>0.05. (q) Statistics of the AO-positive cell number in (k–o). For each genotype, at least 10 discs were analyzed. Three asterisks, P<0.001; n.s., P>0.05
Compared with the loss-of-ACV phenotype produced by ptc>Hep (Figure 3g), ptc>Egr was reported to generate a much severe wing phenotype,32 presumably due to their different abilities to activate Bsk-mediated cell death. Indeed, Egr induced stronger cell death (AO staining) and Bsk activation (puc-LacZ expression) than Hep in the wing disc (Supplementary Figures S3b, e, i and S4).
Wg signaling promotes caspase-independent cell death
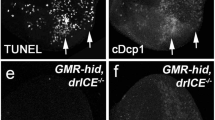
To further explore the role of Wg signaling in cell death, we activated the canonical Wg pathway in the developing eye. Expression of Wg, Dsh, or Arm driven by GMR-Gal4 promotes extensive cell death as revealed by AO staining (Figures 4i–k) and TUNEL assay (Figures 4o–q) that visualizes dying cells,38, 39 and produces eyes with reduced size (Figures 4c–e). All these phenotypes recapitulate that of Bsk activation (Figures 4b, h and n) or expression of the proapoptotic gene Rpr (Figures 4f, l and r). Due to different expression level of each UAS line and the nature of the proteins, the resulted small eye phenotypes exhibit some variation in the size. Consistent with previous study that activated Bsk signaling triggers caspase-independent cell death (Supplementary Figure S5a),32 activation of Bsk or Wg signaling does not elicit caspase activation, as detected by an antibody that specifically recognizes the cleaved caspase-3 (Cas3*) (Figures 4s–w). As a positive control, expression of Rpr results in a very strong Cas3* staining (Figure 4x). Furthermore, gain of Wg signaling-triggered small eye phenotypes (Supplementary Figures S5b–d) are not suppressed by deficiency Df(3 L)H99 (Supplementary Figures S5e–g) that deletes all three proapoptotic genes (rpr, hid, and grim), expression of the caspase inhibitor Diap1 (Supplementary Figures S5h–j), a dominant-negative form or RNAi of the initiator caspapse dronc (Supplementary Figures S5k–p), or knockdown of the effector caspase drice and dcp-1 (Supplementary Figures S5q–v). Intriguingly, expression of the caspase inhibitor P35 mildly suppressed Wg signaling-triggered small eye phenotypes (Supplementary Figures S6a–c and g–i). Thus, P35 may function as a general cell death inhibitor that blocks both caspase-dependent and -independent cell death. Consistently, expression of P35 also suppresses activated Bsk- or P53-induced cell death phenotype (Supplementary Figures S6d–f and j–l). Besides, Wg signaling-triggered small eye phenotypes are not suppressed by expressing two independent dominant-negative Dp53 (Supplementary Figure S7). Together, these data suggest that activation of Wg signaling contributes to Bsk-mediated, but caspase- and Dp53-independent, cell death in vivo.

Wg signaling promotes caspase-independent cell death. Light micrographs of Drosophila adult eyes (a–f) and fluorescent micrographs of third instar eye discs (g–x) are shown. Compared with the GMR-Gal4 control (a, g, m, s), expression of HepCA, Wg, Dsh, or Arm induces reduced-eye size phenotypes (b–e), increased cell death by AO (h–k) and TUNEL staining (n–q), but no caspase activation indicated by cleaved caspase-3(Cas3*) antibody staining (t–w). Expression of Rpr serves as a positive control which induces caspase-dependent cell death (f, l, r, x)
Wg signaling is necessary and sufficient for puc expression
Activation of Bsk results in transcriptional upregulation of puc, which serves as an in vivo readout of Bsk signaling.40 To investigate whether Wg signaling contributes to Bsk-induced puc transcription, we checked the expression of puc-LacZ, a LacZ bearing P-element inserted into the puc locus (also known as pucE69).41 In wild-type wing discs of third instar larva, puc is only expressed in the dorsal tip (Figure 5a). Activation of Bsk signaling along the A/P compartment boundary by ptc>Egr or ptc>Hep induces puc expression (Figure 5b and Supplementary Figure S8b), which is significantly suppressed by RNAi-mediated downregulation of canonical Wg signaling (Figures 5c and d, and Supplementary Figures S8c and d). Additionally, activated Bsk also induces puc expression in the salivary gland, which is suppressed by blocking Wg signaling (Figures 5e–h).

Wg signaling is necessary and sufficient for puc expression. Light micrographs of Drosophila third instar wing discs (a–d, i–l) and salivary glands (e–h, m–p) with X-Gal staining are shown. Compared with the ptc-Gal4 control (a, e), ectopic Egr-induced puc-LacZ expression in wing disc (b) or salivary gland (f) is suppressed by RNAi knocking-down of wg (c), arm (d), or pan (g), or expression of Sgg (h). Compared with the sd-Gal4 (i) and ptc-Gal4 (m) control, ectopic expression of Hep (j, n), Dsh (k), Pan (l, p), or Arm (o) triggers puc-LacZ expression in wing disc (j–l) or salivary gland (n–p)
To examine whether Wg signaling is sufficient to elicit puc expression, we activated the canonical Wg pathway in the wing pouch or salivary gland, driven by sd-Gal4 or ptc-Gal4 respectively. Expression of Hep was included as a positive control. We observed that puc-LacZ expression was dramatically enhanced upon activation of Wg or Bsk signaling (Figures 5i–p). Consistently, the expression of endogenous puc gene is also elevated by activated Wg or Bsk signaling (Supplementary Figure S8j). However, unlike activation of Bsk signaling, activated Wg pathway cannot activate puc expression in the eye discs (Supplementary Figures S8g–i), suggesting that Wg signaling induces puc expression in a context-dependent manner. It is likely that additional factor(s) presented only in the wing disc and salivary gland, but not in the eye disc, is required for Wg signaling to induce puc expression.
Together, the above data provide compelling evidence that the canonical Wg pathway is both necessary and sufficient for Bsk-induced puc expression in both the wing disc and the salivary gland.
Activation of Bsk signaling promotes wg transcription
We have shown that Bsk-mediated cell death requires Wg pathway, which places the Wg signaling downstream of Bsk. Consistent with this explanation, gain of Wg signaling-induced cell death is not suppressed by BskDN (Supplementary Figure S9). Given the fact that mutation or RNAi-mediated downregulation of wg gene abrogates Bsk-mediated cell death (Figures 1d, e, and i and Supplementary Figure S1b), wg expression is probably upregulated by Bsk activation. To test this hypothesis, we first checked Wg protein expression by immunostaining with an anti-Wg antibody. In third instar larva wing discs, endogenous Wg is expressed along the Dorsal/Ventral (D/V) boundary and in two concentric rings in the wing pouch (Figure 6a).16 Overexpression of Egr or HepCA in the posterior compartment of wing disc results in elevated Wg protein level, as compared with that in the anterior compartment (Figures 6b and c). The increased Wg protein level is due to upregulated wg transcription, as revealed by a wg-LacZ reporter (Figures 6d–f). We also noticed that expression of HepCA triggers strong cell death (data not shown) that resulted in a dramatic reduction of the posterior compartment (Figures 6c and f). Thus, we conclude that activation of Bsk signaling is able to promote wg transcription.

Activation of Bsk signaling promotes wg transcription. Fluorescent micrographs of third instar wing pouches are shown. Compared with the en-Gal4 UAS-GFP controls (a, d), Wg (a) or wg-LacZ (d) expression is significantly elevated by ectopic expression of Egr (b, e) or HepCA (c, f) in the posterior compartment (indicated by GFP). For (c) and (f), flies were reared at 18 °C till early third instar larvae, HepCA expression was induced at 29 °C for 21 h before dissecting, Wg and wg-LacZ expression is detected by anti-Wg and anti-β-gal antibody
Wg signaling is required for the physiological functions of Bsk
All the above data demonstrate that Wg signaling is required for ectopically activated Bsk-induced cell death, however, it remains unknown whether Wg pathway also contributes to the physiological function of Bsk signaling. To address this question, we generated puc loss-of-function clones in Drosophila imaginal discs. Previous study suggests that Puc, the Bsk phosphatase, ensures cell viability by restraining basal Bsk signaling, whereas puc mutant clones are frequently eliminated from imaginal discs due to Bsk-mediated cell death.42 Consistent with this notion, we observed that puc loss-of-function clones in wing pouch were considerably smaller than wild-type controls, and could be rescued significantly by blocking Wg signaling (Figures 7a–d). Similar results were also obtained in eye discs (Supplementary Figure S10). Thus, the canonical Wg pathway is critically required for the physiological function of Bsk signaling in regulating epithelia cell viability.

Wg signaling is required for the physiological functions of Bsk. Compared with wild-type clones in the wing pouch (a), puc loss-of-function clones show dramatically reduced size (b), which is fully rescued by RNAi knocking-down of pan (c). (d) Statistics of total clone areas in (a–c). For each genotype, at least 10 clones were analyzed. Three asterisks, P<0.001; n.s., P>0.05. Compared with the ptc-Gal4 control (e), knocking-down of puc along the A/P boundary results in a loss-of-ACV phenotype (f), which is significantly suppressed by knocking-down of wg (g) or expression of Sgg (h). (i) Statistics of the loss-of-ACV phenotype in (e–h) (e, 0.00%, n=301; f, 96.03%, n=126; g, 1.25%, n=240; h, 30.00%, n=100). Three asterisks, P<0.001
Knocking-down of puc along the A/P compartment boundary in developing wings by the ptc-Gal4 driver results in a loss-of-ACV phenotype (Figure 7f). This phenotype resembles that of Bsk activation (Figure 3g), and is significantly rescued by downregulation of Wg pathway (Figures 7g–i), suggesting that Wg pathway contributes to the physiological function of Bsk signaling in vein development.
Loss of cell polarity genes, including scribble (scrib), lethal giant larvae (lgl) and disc large (dlg),43 results in Bsk-mediated cell death and elimination from the tissue.44, 45 Knocking-down of lgl along the A/P compartment boundary (ptc>lgl-IR) in the wing pouch induces Bsk-dependent cell death (Supplementary Figures S11a–d and g), which is significantly suppressed by depletion of wg or dsh (Supplementary Figures S11e–g), suggesting that the Wg pathway is also required for loss-of-cell-polarity induced Bsk-mediated cell death.
Discussion
The interplay between JNK signaling and the non-canonical Wnt/Fz-PCP pathway has been extensively studied, in which JNK acts downstream of Dsh through Rho family small GTPase, and regulates the key effectors such as Myosin II and other cell adhesion components.26, 27, 28 PCP establishment has vital roles in the pattern formation of ommatidia, wing hair and thoracic bristles in Drosophila,27 and in axis elongation, neural tube closure, inner ear pattering and wound healing in vertebrate.46, 47 On the other hand, relative little is known about the crosstalk between JNK and the canonical Wnt/Wg pathways. Previous study has suggested a collaboration between Bsk and the canonical Wg pathway during Drosophila embryogenesis. However, the two signaling routes appear to function in parallel, and the underlying mechanism remains unclear.29 Apoptotic cells, when kept alive ('undead cells') by expressing P35, induce compensatory proliferation in neighbor cells by secreting growth factors like Wg, Dpp and Hh.48, 49, 50 Both secretion of growth factors and compensatory proliferation depends on Bsk signaling,4, 51, 52 yet the mechanism has not been fully illustrated. In mammal, a connection between c-Jun and TCF4, transcription factors of JNK and Wnt pathways respectively, is reported to have a role in intestinal tumorigenesis.30
In the present study, we characterize the genetic interaction between Bsk and the canonical Wg signaling in Drosophila, and obtain the following results: (1) loss of Wg signaling suppresses Bsk-mediated caspase-independent cell death and puc expression; (2) activation of Wg signaling induces caspase-independent cell death and puc expression, which challenges the previous opinion that puc is a direct transcription target and readout of Bsk signaling. We cannot rule out the possibility that a context-dependent feed-back loop may exist between Wg and Bsk signaling; (3) Wg signaling acts downstream of Bsk in promoting cell death; (4) Wg pathway participates in the physiological function of Bsk signaling; (5) activated Bsk signaling results in upregulated wg transcription. Thus, we not only deliver compelling evidences for the conclusion that activated Bsk signaling promotes Wg pathway-dependent cell death, but also provide the underlying mechanism for the interplay of the two pathways that have crucial roles in development. However, since Wg signaling is necessary for cell proliferation and viability, complete loss of Wg signaling would result in developmental defect and animal lethality, we used heterozygous mutants or RNAi-mediated knock-down approach to examine the effect of loss-of-Wg signaling on Bsk-mediated cell death. In such backgrounds, Wg signaling is effectively reduced, but not completely abolished. Consequently, Bsk-mediated cell death and resulted phenotypes are significantly, but not fully, suppressed (Figures 1, 2, 3). Therefore, we could not exclude the possibility that other factor(s) or signaling pathway(s) may exist in parallel with Wg signaling to mediate Bsk signaling-induced cell death (Supplementary Figure S5a).
As an evolutionary conserved signaling pathway, Drosophila Wg signaling has been shown to have important roles for developmentally regulated cell death, like sculpting the retina and ommatidia.53, 54, 55 Previous works suggested that Wg signaling induces ommatidial elimination through elevated expression of pro-apoptotic factors rpr, hid and grim.54 In contrast with the finding, we show Wg signaling induces caspase-independent cell death, evident by the fact that no cleaved caspase 3 is detected by antibody staining (Figures 4u–w), and Wg signaling-triggered cell death are not suppressed by Df(3L)H99 and expression of Diap1, a dominant-negative form or knockdown of dronc, or knockdown of drice and dcp-1 (Supplementary Figures S5b–v). One plausible explanation for this discrepancy is that Wg signaling may promote cell death via distinct mechanisms in a context-dependent manner. Since Bsk is also involved in necroptosis triggered by factors such as the mitochondrial protein apoptosis-inducing factor (AIF),56, 57 it would be interesting to investigate in future whether Wg signaling-induced caspase-independent cell death has a role in necroptosis.
Since the discovery of the first Wnt gene more than 30 years ago,58 research on Wnt signaling has growing into one of the most active field-, and huge amount of progress have been achieved at an accelerating pace.59 Given the numerous functions of Wnt signaling during development, it is not surprising that deregulation of this pathway is a prevalent theme in cancer biology, especially colorectal cancer (CRC). Constitutively active Wnt signaling has been associated with tumor progression in many cancers, yet in this study we characterize a role of Wg signaling in promoting cell death, which implies that Wnt signaling may also possess a tumor suppressor function. Meanwhile, a substantial body of evidence also suggests that JNK signaling is closely related with tumor formation and metastasis,60, 61, 62, 63, 64 thus the present study shed new light on the crosstalk and involvement of JNK and Wnt signaling in cell death and cancer development.
Materials and Methods
Drosophila strains and generation of clones
The following stocks have been described previously: wg1–17;65 Sp;34 dsh6;66 wgIG22, dshV26, armXM19;67 arm1, pan13a;68 SggEP1576;69 UAS-Wg, UAS-Dsh, UAS-wg-IR and UAS-arm-IR;70 UAS-Pan;71 bsk1; 72 UAS-EgrRegg1;10 UAS-HepCA, UAS-Egr, UAS-BskDN, UAS-Puc, Df(3 L)H99, DIAP1, DRONCDN and GUS-Dp53259H;32 UAS-HepWT;31 pucE69, pnr-Gal4;12 GMR-Gal4, ptc-Gal4 and sd-Gal4;73 UAS-Dp53H159N.35 The wg-LacZ, UAS-Rpr, UAS-Dp53, UAS-Arm, UAS-Apc2, UAS-Axn, and UAS-dsh-IR lines were obtained from the Bloomington stock center. The UAS-pan-IR and UAS-lgl-IR lines were obtained from the Vienna Drosophila RNAi Center (VDRC). The UAS-wg-IR, UAS-arm-IR, UAS-dronc-IR, UAS-drice-IR, UAS-dcp-1-IR lines were got from Fly Stocks of National Institute of Genetics (NIG-FLY).
Fluorescently labelled clones were produced in larval imaginal discs using the y w1118 hs-Flp; act>CD2>Gal4 UAS-GFP/Cyo strain. Clones were induced by heat shock at 37 °C for 10 min, late third-instar larvae were dissected after recovering for 3 days.
Immunostaining
The following primary antibodies were used: 1 : 400 rabbit-anti-Caspase 3 (Cell Signaling Technology, CST, Cat # 9661, Danvers, MA, USA), 1 : 300 mouse-anti-Wingless (Developmental Studies Hybridoma Bank, DSHB, lova City, IA, USA), 1 : 500 mouse-anti-β-Gal (DSHB). The second antibodies were used as follows: 1 : 1000 anti-mouse CY3 (CST), 1 : 1000 anti- rabbit CY3 (CST).
AO staining
The discs were dissected at the third instar larva stage, and stained for AO as described.32
TUNEL staining
The discs were dissected at the third instar larva stage, and stained for TUNEL using the Fluorescein Cell Death Kit produced by Boster Company. Imaging of prepared sample was conducted by a Leica confocal microscope (Leica SP5, Solms, Germany).
X-Gal staining
The discs were dissected at the third instar larva stage, and stained for β-galactosidase as described.74
qRT-PCR
Wing discs were dissected at third instar larva, for each genotype, more than 200 discs were collected and total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA). qRT-PCR was performed as previously described,75 primers pairs of rp4975 and puc76 were used as previously described.
Statistics
For loss of ACV phenotype, statistics were analyzed using chi-square test. For AO stanining and area of disc clones, one-way analysis of variance test followed by the post Dunnett test or Kruskal–Wallis text followed by the post Dunns text was used. A P-value of <0.05 was considered as significant.
Abbreviations
- JNK:
-
c-Jun N-terminal kinase
- TNF:
-
tumor necrosis factor
- APC:
-
adenomatous polyposis coli protein
- CRC:
-
colorectal cancer
- PCP:
-
planar cell polarity
- GMR:
-
glass multiple reporter
- ACV:
-
anterior cross vein
- A/P:
-
anterior/posterior
- D/V:
-
dorsal/ventral
- AO:
-
acridine orange
- RNAi:
-
RNA interference
References
Baehrecke EH . How death shapes life during development. Nat Rev Mol Cell Biol 2002; 3: 779–787.
Conradt B . Genetic control of programmed cell death during animal development. Annu Rev Genet 2009; 43: 493–523.
Yuan J, Yankner BA . Apoptosis in the nervous system. Nature 2000; 407: 802–809.
Fuchs Y, Steller H . Programmed cell death in animal development and disease. Cell 2011; 147: 742–758.
Igaki T, Miura M . The Drosophila TNF ortholog Eiger: emerging physiological roles and evolution of the TNF system. Semin Immunol 2014; 26: 267–274.
Weston CR, Davis RJ . The JNK signal transduction pathway. Curr Opin Cell Biol 2007; 19: 142–149.
Weston CR, Davis RJ . The JNK signal transduction pathway. Curr Opin Genet Dev 2002; 12: 14–21.
Luo X, Puig O, Hyun J, Bohmann D, Jasper H . Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J 2007; 26: 380–390.
Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 2004; 23: 4802–4812.
Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 2002; 21: 3009–3018.
Moreno E, Yan M, Basler K . Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 2002; 12: 1263–1268.
Xue L, Igaki T, Kuranaga E, Kanda H, Miura M, Xu T . Tumor suppressor CYLD regulates JNK-induced cell death in Drosophila. Dev Cell 2007; 13: 446–454.
Igaki T . Correcting developmental errors by apoptosis: lessons from Drosophila JNK signaling. Apoptosis 2009; 14: 1021–1028.
Ma X, Li W, Yu H, Yang Y, Li M, Xue L et al. Bendless modulates JNK-mediated cell death and migration in Drosophila. Cell Death Differ 2014; 21: 407–415.
Miller JR . The Wnts. Genome Biol 2014; 3: REVIEWS3001.
Swarup S, Verheyen EM . Wnt/Wingless in Drosophila. Cold Spring Harb Perspect Biol 2012; 4: 1–15.
MacDonald BT, Tamai K, He X . Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26.
Nusse R . Wnt signaling in disease and in development. Cell Res 2005; 15: 28–32.
Reya T, Clevers H . Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–850.
Clevers H, Nusse R . Wnt/beta-catenin signaling and disease. Cell 2012; 149: 1192–1205.
Sharma RP, Chopra VL . Effect of the Wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev Biol 1976; 48: 461–465.
Baker NE . Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: the spatial distribution of a transcript in embryos. EMBO J 1987; 6: 1765–1773.
Rijsewijk F, Schuermann M, Wagenaar E, Parren P, Weigel D, Nusse R . The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987; 50: 649–657.
Bejsovec A . Flying at the head of the pack: Wnt biology in Drosophila. Oncogene 2006; 25: 7442–7449.
Baarsma HA, Konigshoff M, Gosens R . The WNT signaling pathway from ligand secretion to gene transcription: molecular mechanisms and pharmacological targets. Pharmacol Ther 2013; 138: 66–83.
Singh J, Mlodzik M . Planar cell polarity signaling: coordination of cellular orientation across tissues. Wiley interdisciplinary reviews. Dev Biol 2012; 1: 479–499.
Maung SM, Jenny A . Planar cell polarity in Drosophila. Organogenesis 2011; 7: 165–179.
Fanto M, McNeill H . Planar polarity from flies to vertebrates. J Cell Sci 2004; 117: 527–533.
McEwen DG, Cox RT, Peifer M . The canonical Wg and JNK signaling cascades collaborate to promote both dorsal closure and ventral patterning. Development 2000; 127: 3607–3617.
Saadeddin A, Babaei-Jadidi R, Spencer-Dene B, Nateri AS . The links between transcription, beta-catenin/JNK signaling, and carcinogenesis. Mol Cancer Res 2009; 7: 1189–1196.
Ma X, Yang L, Yang Y, Li M, Li W, Xue L . dUev1a modulates TNF-JNK mediated tumor progression and cell death in Drosophila. Dev Biol 2013; 380: 211–221.
Ma X, Huang J, Yang L, Yang Y, Li W, Xue L . NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res 2012; 22: 425–431.
Lindsley DL, Zimm GG . The Genome of Drosophila melanogaster. Academic Press: San Diego, 1992.
Neumann CJ, Cohen SM . Sternopleural is a regulatory mutation of wingless with both dominant and recessive effects on larval development of Drosophila melanogaster. Genetics 1996; 142: 1147–1155.
Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 2000; 101: 91–101.
Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM . Drosophila p53 binds a damage response element at the reaper locus. Cell 2000; 101: 103–113.
Fan Y, Lee TV, Xu D, Chen Z, Lamblin AF, Steller H et al. Dual roles of Drosophila p53 in cell death and cell differentiation. Cell Death Differ 2010; 17: 912–921.
Abrams JM, White K, Fessler LI, Steller H . Programmed cell death during Drosophila embryogenesis. Development 1993; 117: 29–43.
Charroux B, Royet J . Elimination of plasmatocytes by targeted apoptosis reveals their role in multiple aspects of the Drosophila immune response. Proc Natl Acad Sci USA 2009; 106: 9797–9802.
Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 1998; 12: 557–570.
Agnes F, Suzanne M, Noselli S . The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 1999; 126: 5453–5462.
McEwen DG, Peifer M . Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 2005; 132: 3935–3946.
Bilder D, Perrimon N . Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature 2000; 403: 676–680.
Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T . Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev Cell 2009; 16: 458–465.
Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, Igaki T . Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev Cell 2011; 20: 315–328.
Mayor R, Theveneau E . The role of the non-canonical Wnt-planar cell polarity pathway in neural crest migration. Biochem J 2014; 457: 19–26.
Wansleeben C, Meijlink F . The planar cell polarity pathway in vertebrate development. Dev Dyn 2011; 240: 616–626.
Ryoo HD, Gorenc T, Steller H . Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 2004; 7: 491–501.
Perez-Garijo A, Martin FA, Morata G . Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 2004; 131: 5591–5598.
Huh JR, Guo M, Hay BA . Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol 2004; 14: 1262–1266.
Martin FA, Perez-Garijo A, Morata G . Apoptosis in Drosophila: compensatory proliferation and undead cells. Int J Dev Biol 2009; 53: 1341–1347.
Morata G, Shlevkov E, Perez-Garijo A . Mitogenic signaling from apoptotic cells in Drosophila. Dev Growth Differ 2011; 53: 168–176.
Cordero J, Jassim O, Bao S, Cagan R . A role for wingless in an early pupal cell death event that contributes to patterning the Drosophila eye. Mech Dev 2004; 121: 1523–1530.
Lin HV, Rogulja A, Cadigan KM . Wingless eliminates ommatidia from the edge of the developing eye through activation of apoptosis. Development 2004; 131: 2409–2418.
Cordero JB, Cagan RL . Canonical wingless signaling regulates cone cell specification in the Drosophila retina. Dev Dyn 2010; 239: 875–884.
Yuan J, Kroemer G . Alternative cell death mechanisms in development and beyond. Genes Dev 2010; 24: 2592–2602.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature reviews. Mol Cell Biol 2010; 11: 700–714.
Nusse R, Varmus HE . Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982; 31: 99–109.
Nusse R, Varmus H . Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J 2012; 31: 2670–2684.
Kennedy NJ, Davis RJ . Role of JNK in tumor development. Cell Cycle 2003; 2: 199–201.
Wagner EF, Nebreda AR . Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9: 537–549.
Chen F . JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res 2012; 72: 379–386.
Sabapathy K . Role of the JNK pathway in human diseases. Prog Mol Biol Transl Sci 2012; 106: 145–169.
Davies C, Tournier C . Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans 2012; 40: 85–89.
Yang M, Hatton-Ellis E, Simpson P . The kinase Sgg modulates temporal development of macrochaetes in Drosophila by phosphorylation of Scute and Pannier. Development 2012; 139: 325–334.
Vandewalle J, Langen M, Zschätzsch M, Nijhof B, Kramer JM, Brems H et al. Ubiquitin ligase HUWE1 regulates axon branching through the Wnt/beta-catenin pathway in a Drosophila model for intellectual disability. PLoS ONE 2013; 8: e81791.
Tolwinski NS, Wieschaus E . A nuclear function for armadillo/beta-catenin. PLoS Biol 2004; 2: E95.
Helms W, Lee H, Ammerman M, Parks AL, Muskavitch MA, Yedvobnick B . Engineered truncations in the Drosophila mastermind protein disrupt Notch pathway function. Dev Biol 1999; 215: 358–374.
Singh A, Chan J, Chern JJ, Choi KW . Genetic interaction of Lobe with its modifiers in dorsoventral patterning and growth of the Drosophila eye. Genetics 2005; 171: 169–183.
Li WZ, Li SL, Zheng HY, Zhang SP, Xue L . A broad expression profile of the GMR-GAL4 driver in Drosophila melanogaster. Genet Mol Res 2012; 11: 1997–2002.
Kiger JA Jr, Natzle JE, Green MM . Hemocytes are essential for wing maturation in Drosophila melanogaster. Proc Natl Acad Sci USA 2001; 98: 10190–10195.
Takatsu Y, Nakamura M, Stapleton M, Danos MC, Matsumoto K, O'Connor MB et al. TAK1 participates in c-Jun N-terminal kinase signaling during Drosophila development. Mol Cell Biol 2000; 20: 3015–3026.
Wang X, Wang Z, Chen Y, Huang X, Hu Y, Zhang R et al. FoxO mediates APP-induced AICD-dependent cell death. Cell Death Dis 2014; 5: e1233.
Xue L, Noll M . Dual role of the Pax gene paired in accessory gland development of Drosophila. Development 2002; 129: 339–346.
Beck ES, Gasque G, Imlach WL, Jiao W, Jiwon Choi B, Wu PS et al. Regulation of Fasciclin II and synaptic terminal development by the splicing factor beag. J Neurosci 2012; 32: 7058–7073.
Kim JH, Wang X, Coolon R, Ye B . Dscam expression levels determine presynaptic arbor sizes in Drosophila sensory neurons. Neuron 2013; 78: 827–838.
Acknowledgements
We thank Drs Zhaohui Wang, Xinhua Lin, the Bloomington Drosophila Stock Center, the Vienna Drosophila RNAi Center and National Institute of Genetics (NIG) for fly stocks, members of Xue lab for discussion and critical comments. This work is supported by the National Basic Research Program of China (973 Program) (2011CB943903), National Natural Science Foundation of China (31071294, 31171413, 31371490), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20120072110023), and Shanghai Committee of Science and Technology (09DZ2260100, 14JC1406000).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, S., Chen, C., Wu, C. et al. The canonical Wg signaling modulates Bsk-mediated cell death in Drosophila. Cell Death Dis 6, e1713 (2015). https://doi.org/10.1038/cddis.2015.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.85
This article is cited by
-
Slik maintains tissue homeostasis by preventing JNK-mediated apoptosis
Cell Division (2023)
-
Dysfunction of lipid storage droplet-2 suppresses endoreplication and induces JNK pathway-mediated apoptotic cell death in Drosophila salivary glands
Scientific Reports (2022)
-
Global chromatin organizer SATB1 acts as a context-dependent regulator of the Wnt/Wg target genes
Scientific Reports (2021)
-
Snail modulates JNK-mediated cell death in Drosophila
Cell Death & Disease (2019)
-
ADAMTS Sol narae cleaves extracellular Wingless to generate a novel active form that regulates cell proliferation in Drosophila
Cell Death & Disease (2019)
