
Abstract
We previously reported that IL-2 deprivation induced acid sphingomyelinase-mediated (ASM-mediated) ceramide elevation and apoptosis in an NK/T lymphoma cell line KHYG-1. However, the molecular mechanism of ASM–ceramide-mediated apoptosis during IL-2 deprivation is poorly understood. Here, we showed that IL-2 deprivation induces caspase-dependent apoptosis characterized by phosphatidylserine externalization, caspase-8, -9, and -3 cleavage, and degradation of X-linked inhibitor of apoptosis protein (XIAP). IL-2 re-supplementation rescued apoptosis via inhibition of XIAP degradation without affecting caspase cleavage. However, IL-2 deprivation induced ceramide elevation via ASM in lysosomes and activated lysosomal cathepsin B (CTSB) but not cathepsin D. A CTSB inhibitor CA-074 Me and knockdown of CTSB inhibited ceramide-mediated XIAP degradation and apoptosis. Inhibition of ceramide accumulation in lysosomes using an ASM inhibitor, desipramine, decreased cytosolic activation of CTSB by inhibiting its transfer into cytosol from the lysosome. Knockdown of ASM also inhibited XIAP degradation and apoptosis. Furthermore, cell permeable N-acetyl sphingosine (C2-ceramide), which increases mainly endogenous d18:1/16:0 and d18:1/24:1 ceramide-like IL-2 deprivation, induced caspase-dependent apoptosis with XIAP degradation through CTSB. These findings suggest that lysosomal ceramide produced by ASM mediates XIAP degradation by activation of cytosolic CTSB and caspase-dependent apoptosis. The ASM–ceramide–CTSB signaling axis is a novel pathway of ceramide-mediated apoptosis in IL-2-deprived NK/T lymphoma cells.
Similar content being viewed by others

Main
KHYG-1 natural killer T (NK/T) cells established from a patient with aggressive NK/T cell lymphoma displayed morphology of a large granular lymphocyte with a large nucleus, rough chromatin, and bulk basophilic cytoplasm.1 Suck et al.2 reported that KHYG-1 cells had in vitro cytotoxicity against lymphoma cell lines such as HL-60 and induced apoptosis of tumor cells via the granzyme M/perforin pathway. Thus KHYG-1 cells have a cytotoxic ability similar to NK cells against malignant cells. However, because interleukin-2 (IL-2) is essential for clonal expansion of KHYG-1 cells, this cell line is a useful model to investigate the mechanism by which NK/T lymphoma cells undergo programmed cell death. Previously, we reported that IL-2 deprivation (IL-2(−)) promoted ceramide generation due to the activation of acid sphingomyelinase (ASM), resulting in apoptosis of KHYG-1 cells.3 The mechanisms of ASM–ceramide-mediated apoptotic signal in IL-2 deprived NK/T lymphoma cells has not been clarified.
Apoptosis uses two major signaling pathways to induce programmed cell death; extrinsic and intrinsic pathways.4 The extrinsic pathway is mediated by extracellular death ligands (Fas ligand, tumor necrosis factor-α (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL), or CD95) and activates death-inducing signaling complex-containing caspase-8 through those receptors. The intrinsic pathway mediates the disruption of mitochondria and induced the formation of an apoptosome complex composed of cytochrome C secreted from mitochondria, apoptotic protease-activating factor 1 (Apaf-1), and caspase-9.4 Both extrinsic and intrinsic pathways lead to the activation of caspase-3/-7, which trigger various apoptotic phenomena such as phosphatidylserine (PS) externalization or DNA fragmentation. In addition to these mechanisms that induce apoptosis, it is important to clarify the role of lysosomal proteases in the regulation of antiapoptotic proteins such as Bcl-2 family members and pro-apoptotic proteins such as caspases.
Cathepsins (CTSs) in lysosomes consist of cysteine protease, aspartic protease cathepsin D (CTSD) and serine protease cathepsin B (CTSB). Upon extracellular and intracellular stressors, CTSs are released into the cytosol and are activated enzymatically by optimal pH conditions.5 Apoptotic signals mainly inhibit antiapoptotic molecules such as Bcl-2 to activate pro-apoptotic Bax/Bak molecules by their degradation through CTSs.6, 7 Currently, the role of CTSD and CTSB in apoptosis is controversial. The activation of CTSB through transforming growth factor-β signaling was reported to increase the proliferation of melanoma cells and short hairpin RNA (shRNA) of CTSB had an apoptotic effect mediated through the degradation of X-linked inhibitor of apoptosis protein (XIAP) in invasive meningioma cells, suggesting the positive effect of CTSB in cell proliferation.8, 9 In contrast, it was reported that CTSB induced apoptosis by activating caspase-3 and -9 in dengue virus-infected HepG2 hepatocytes.10
Caspase-3, -7, and -9 are inhibited by XIAP, an IAP family member11, 12 that directly binds to and inactivates caspase-3 or caspase-9 to inhibit their degradation, resulting in suppression of apoptosis.13, 14, 15, 16 Downregulation of XIAP increases the sensitivity of cancer cells to apoptotic stimuli, such as TRAIL or hypoxia.17, 18 In hematological malignancies, anti-CD33 antibodies induced apoptosis by decreasing XIAP in acute myeloid lymphoma (AML),19 and AML patients with overexpression of XIAP showed unfavorable responses to induction chemotherapy.20 Anticancer drug-resistant lymphoma cells also had overexpression of XIAP through the NF-κB-dependent MEK/MAPK pathway.21 In general, cytosolic proteins such as XIAP are regulated at the transcriptional level or by enzymatic degradation via proteases. However, the molecular mechanism by which the protein levels of XIAP are regulated in ceramide-induced NK/T cell apoptosis has not been investigated.
Ceramide is at the center of sphingolipid metabolism and acts as a substrate of other sphingolipids, such as sphingomyelin, sphingosine-1-phosphate (S1P), and glycosphingolipids.22, 23 Ceramide is also a lipid mediator that induces programmed cell death, differentiation, senescence, cell cycle arrest, and autophagy.22, 23, 24 There are three pathways in intracellular ceramide generation: (i) de novo synthesis from L-serine and a palmitoyl-coenzyme A, (ii) the sphingomyelin cycle consisting of sphingomyelin synthase (SMS) and sphingomyelinase, and (iii) the salvage pathway where ceramide synthases utilize sphingosine degraded from SM, glycolipids, and S1P as a substrate of ceramide.22 These pathways are mutually involved in the generation of ceramide induced by various stimuli.23 Especially, ASM-generated ceramide has been well investigated in numerous types of cell death. Stimulation of TRAIL or CD95 ligands induces rapid ASM activation and formation of ceramide-enriched platforms in the plasma membrane.25, 26 ASM-generated ceramide provides a place to form clusters between ligands and their transmembrane receptors, which transduce an efficient death signal to the intracellular compartment.25, 26, 27 However, stimulation of TNF-α or gemcitabine generated ceramide in lysosomes through ASM activation. Lysosomal ceramide was reported to trigger the CTSD-mediated apoptotic pathway.28, 29, 30 Recently, arsenic trioxide induced the degradation of XIAP through the ubiquitin–proteasome pathway and treatment with valproic acid increased CTSB-induced apoptosis of chronic lymphoid lymphoma cells.31, 32
However, how ASM-generated lysosomal ceramide is related to the cathepsin family, including CTSB and CTSD and XIAP in NK/T lymphoma cell apoptosis, is poorly understood. In this study, we demonstrated that IL-2(−) activated the ASM–ceramide pathway in lysosomes and that generation of d18:1/16:0 and d18:1/24:1 lysosomal ceramides caused the release of CTSB, but not CTSD, into the cytosol. CTSB-mediated degradation of XIAP subsequently induced activation of caspase-3 and its nuclear entry to execute apoptosis. These results suggest the ASM/ceramide/CTSB axis is a novel pathway for the degradation of XIAP in IL-2-deprived NK/T lymphoma cell apoptosis.
Results
IL-2 rescued cells from caspase-dependent apoptosis without inhibition of caspase-3 cleavage in IL-2(−) condition
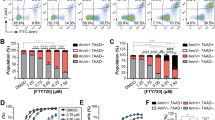
We previously showed that IL-2(−) induced ceramide accumulation that mediated cell death with DNA condensation in KHYG-1 cells.3 The cells were incubated for 12 h in IL-2(−) and then untreated or IL-2 was added to the media (indicated as 0 h in Figure 1a). After 24 h of IL-2(−), apoptotic cells were detected with flow cytometry analysis of Annexin V, a marker of PS externalization and caspase-dependent apoptosis (Figure 1b). Annexin V-positive cells were increased in IL-2(−) compared with IL-2 containing medium (IL-2(+)). We confirmed our previous data that IL-2 supplementation (rescue) could prevent cells undergoing IL-2(−)-mediated apoptosis (Figure 1b). To examine caspase activation, we performed in vivo casapse-3/7 activity in KHYG-1 cells (Figure 1c). After 12 h, IL-2(−) induced activation of caspase-3/7, but IL-2 rescue inhibited its activation, suggesting IL-2(−) induced caspase-dependent apoptosis, and IL-2 rescue could rescue apoptosis. Pro-caspases are cleaved and the cleaved form activates apoptotic signal. As shown Figure 1d, cleaved caspase-3 was observed with nuclear condensation in some IL-2(−)-treated cells compared with IL-2(+)-treated cells. Interestingly, cleaved caspase-3 was also detected without nuclear condensation and localized in cytosol in rescued cells (Figure 1d). To confirm caspase cleavage in IL-2(−) and rescue cells, we performed western blotting analysis. Pro-caspase-3 (32 kD), pro-caspase-8 (55 kD), and pro-caspase-9 (42 kD) were cleaved by IL-2(−) treatment (Figure 1e). According to caspase cleavage, poly ADP ribose polymerase (PARP), a caspase-3 target and marker of apoptosis, was cleaved by IL-2(−) (Figure 1e). However, in rescue cells, PARP cleavage (86 kD) was not observed, even though caspases were cleaved (Figure 1e). These results suggested that IL-2(−) induced caspase activation and subsequent apoptosis, and rescue inhibited apoptosis through a mechanism involving inhibition of caspase activity but not caspase cleavage. Indeed, a caspase inhibitor, DEVD-CHO, inhibited IL-2(−)-induced cell death by a concentration-dependent mechanism (Figure 1f).

IL-2(−) induced caspase-dependent apoptosis. (a) Time schedule of study. KHYG-1 cells were cultured in RPMI1640 containing 10% FCS with (IL-2(+)) or without (IL-2(−)) IL-2 (100 U/ml). After 12 h of IL-2 deprivation, IL-2 (100 U/ml) was added to media (rescue), and this point was indicated as 0 h. (b) Cells were cultured at each condition for 24 h and stained with fluorescein isothiocyanate (FITC)–Annexin V. Percentage of FITC–Annexin V-positive cells were measured by flow cytometry. (c) In vivo caspase-3/-7 activity was performed with fluorescent substrates of caspase-3/7 (FAM-DEVD-FMK). Cells were cultured at each condition for 36 h. Images were obtained by fluorescent microscopy. The value is mean±S.D. from three different experiments. *P<0.005. (d) Cells were treated with IL-2(+), IL-2(−), and rescue for 12 h, and stained with anticleaved caspase-3 p17 antibody and DAPI. Images were obtained by confocal microscopy. Scale bar, 10 μm. (e) The cells were incubated in IL-2(−) or rescue for the indicated times, and each protein was detected by immunoblotting. (f) Cells were incubated in IL-2(−) at each concentration of DEVD-CHO for 36 h. Surviving cells were counted with trypan blue and indicated as a percentage of 0 h. Values are mean±S.D. from three different experiments. *P<0.005 vs 0 h; **P<0.05 vs 0 h
Ceramide accumulated at lysosomes via ASM activation in IL-2(-)-treated cells
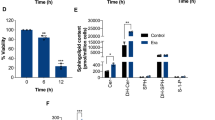
Previously, we showed that ceramide generation in IL-2(−) is important for induction of apoptosis.3 We confirmed that IL-2(−) activated ASM but not neutral sphingomyelinase (NSM), enzymes that generate ceramide from sphingomyelin (Figures 2a and b). According to enhanced ASM activity, ceramide species such as d18:1/16:0, d18:1/18:0, and d18:1/24:1 were significantly increased by IL-2(−) (Figure 2c) but inhibited by IL-2 rescue (Figures 2a–c). ASM acts at the lysosome and outer leaflet of the plasma membrane after activation.33 Thus we assessed where ceramide was produced and accumulated by IL-2(−). As shown in Figure 2d, ceramide was increased and colocalized with Lamp1, a lysosomal marker, in IL-2(−) conditions. IL-2 rescue suppressed ceramide accumulation (Figure 2d). To elucidate accumulation of ceramide in lysosome, we performed lysosome fractionation after treatment with IL-2(+) or IL-2(−) (Figure 2e). As shown in Figures 2f and g, lysosomal ceramide was increased by IL-2(−). In addition, the species of increased ceramide (d18:1/16:0, d18:1/18:0, and d18:1/24:1) in lysosome were same as those in whole cells (Figures 2c and g). These data suggested that IL-2(−) induced lysosomal ceramide accumulation through activation of ASM and inhibition of SMS and glucosylceramide synthase and that lysosomal ceramide was related to IL-2(−)-induced apoptosis.

IL-2(−) induced accumulation of lysosomal ceramide. (a and b) KHYG-1 cells were cultured in IL-2(+), IL-2(−), and rescue conditions for 12 h. Cells were harvested, and after extraction of the proteins, the activities of each enzyme were assessed. Activities of ASM (a) and NSM (b) were measured using C6-NBD-ceramide and C6-NBD-SM as substrates. (c) After 36 h, ceramide levels were assessed by LC-MS/MS. (d) Cells were cultured at each condition for 12 h, fixed, stained with anti-ceramide and Lamp1 (lysosome) antibodies, and analyzed by confocal microscopy. Nuclei were counter-stained with DAPI. Scale bars, 10 μm. (e) After 24 h, lysosome fraction were isolated as described in Materials and Methods section. Lamp2 (lysosome), pan-cadherin (plasma membrane), and GAPDH (cytosol) were used as markers for each organelle in whole cell lysate (W) and lysosome fraction (L). (f and g) Total ceramide levels in whole cell and lysosome (f) and ceramide species in lysosome (g) were assessed by LC-MS/MS. Values are mean±S.D. from three different experiments. *P<0.005, **P<0.05
IL-2(-) treatment induces XIAP degradation
To assess the mechanism that IL-2 rescue inhibits the activity of cleaved caspase, we focused on XIAP, an IAP with inhibitory effects on caspases.11, 12 XIAP inhibits both caspase-3 and caspase-9 through direct binding.13, 14, 15, 16 In the above data, cleavage of caspases was observed in IL-2 rescue without apoptosis. Thus we examined whether XIAP was related to IL-2(−) and rescue. XIAP proteins were decreased by IL-2(−) treatment and IL-2 rescue recovered XIAP levels (Figure 3a). IL-2(−) treatment promoted reduced XIAP levels from 3 h and after 24 h XIAP protein was not detectable (Figure 3b). To confirm XIAP degradation, we observed the localization of XIAP and cleaved caspase-3 by immunocytochemistry and confocal microscopy. As shown in Figure 3d, IL-2(-) treatment decreased XIAP, and cleaved caspase-3 accumulated in the nuclei. However, XIAP colocalized with cleaved caspase-3 in the cytosol by IL-2 rescue (Figure 3d). These data suggested IL-2(−) activates both capase-3 cleavage and XIAP degradation, and IL-2 rescue prevents XIAP degradation but not caspase-3 activation.

Involvement of XIAP degradation on caspase-dependent apoptosis during IL-2(−). (a) KHYG-1 cells were incubated in IL-2(+), IL-2(−), and rescue conditions for 12 h. XIAP and actin were detected by western blotting analysis. (b) Cells were harvested at the indicated times after IL-2(−) treatment. Immunoblotting was performed with anti-XIAP and anti-actin antibodies. (c) Cells were incubated at each condition for 12 h, fixed, stained with XIAP and cleaved caspase-3 antibodies, and analyzed by confocal microscopy. Scale bars, 10 μm
CTSB inhibitor blocks XIAP degradation without ceramide accumulation in IL-2(−)
The above data suggested that lysosomal ceramide induced XIAP degradation and apoptosis during IL-2(−). Thus we examined whether the lysosomal apoptotic pathway is involved in IL-2(−)-induced apoptosis. Lysosomal protease CTSs such as CTSB and CTSD are well-known apoptosis inducers.34 Especially, CTSD is downstream of ceramide.28, 29, 35 To assess the relationship between the cathepsin pathway and IL-2(−)-induced XIAP degradation and apoptosis, we used cathepsin inhibitors. Pepstatin A, a CTSD inhibitor, had no effect on IL-2(−)-induced XIAP degradation (Figure 4a). Interestingly, a CTSB-specific inhibitor, CA-074 Me, prevented XIAP reduction similar to IL-2 rescue (Figure 4a). Next we demonstrated that IL-2-(−) activated CTSB in vivo, which was inhibited by CA-074 Me but not by pepstatin A treatment (Figure 4b).

Involvement of CTSB in IL-2(−)-mediated apoptosis. KHYG-1 cells were treated with IL-2(−) with or without 4 μM cathepsin inhibitors. (a) After 24 h of treatment, western blotting analysis was performed by using anti-XIAP antibody or anti-actin antibody. (b) After 12 h, in vivo CTSB activity was measured. Images were obtained by fluorescent microscopy. Values are mean±S.D. from three different experiments. *P<0.005 vs IL-2(−) with vehicle treatment. (c) After 12 h, cells were fixed and stained with anti-ceramide and anti-Lamp1 antibodies. Nuclei were counter-stained with DAPI. Images were obtained with confocal microscopy. Scale bars, 10 μm. (d) After 24 h of treatment, viable cell numbers were counted by trypan blue staining and indicated as a percentage of IL-2(+). Values are mean±S.D. from three different experiments. *P<0.005 vs IL-2(−) with vehicle treatment. (e) Establishment of control and CTSB knockdown cells by using lentiviral particles of control shRNA (shSCR) and CTSB-specific shRNA (shCTSB). CTSB, CTSD, and actin were assessed by western blotting analysis. (f) After 24 h of treatment, each protein was detected by immunoblotting. (g) After 36 h, viable cell numbers were counted after trypan blue staining and indicated as a percentage of IL-2(+) in each cell. Values are mean±S.D. from four different experiments. **P<0.05
To elucidate whether CTSB activation was downstream of increased lysosomal ceramide, we observed ceramide accumulation in IL-2(−) conditions with or without CA-074 Me treatment. As shown in Figure 4c, CA-074 Me did not prevent lysosomal ceramide accumulation. However, CA-074 Me treatment inhibited apoptosis (Figure 4d). To confirm the effects of CA-074 Me, we performed knockdown of CTSB. KHYG-1 cells were quite difficult to transduce genes transiently. Thus we established CTSB knockdown cell lines by using CTSB-specific shRNA lentivirus (Figure 4e). CTSB knockdown (shCTSB) blocked XIAP degradation and PARP cleavage induced by IL-2(−) (Figure 4f). In addition, cell survival was also increased in shCTSB cells compared with control cells (shSCR) (Figure 4f). These data suggested that accumulation of lysosomal ceramide during IL-2(−) activated CTSB but not CTSD and the subsequent apoptosis.
ASM inhibitor suppressed ceramide accumulation and CTSB-mediated apoptosis
The above data showed that IL-2(−) activated ASM, which induced lysosomal ceramide accumulation. We tested the effects of an ASM inhibitor on IL-2(−)-mediated apoptosis. Preincubation with ASM inhibitor desipramine blocked ASM activation but not NSM during IL-2(−) (Figures 5a and b). Next, we observed IL-2(−)-induced lysosomal ceramide accumulation in the presence or absence of desipramine by immunocytochemistry. As shown in Figure 5c, ceramide accumulation induced by IL-2(−) was suppressed by desipramine treatment. Moreover, desipramine also blocked CTSB activation, which is downstream of lysosomal ceramide (Figure 5d).

ASM inhibitor suppressed CTBS-mediated apoptosis. KHYG-1 cells were treated with IL-2(−) with or without ASM inhibitor, desipramine (des). (a and b) After 12 h, cells were harvested, and after extraction of the proteins, the activities of ASM (a) and NSM (b) were assessed using C6-NBD-SM as substrates. Values are mean±S.D. from three different experiments. *P<0.005. (c) After 12 h, cells were fixed and stained with anti-ceramide and anti-Lamp1 antibodies. Images were obtained with confocal microscopy. Scale bars, 10 μm. (d) After 12 h, in vivo CTSB activity was performed. Images were obtained by using fluorescent microscopy. Values are mean±S.D. from three different experiments. *P<0.005, **P<0.05. (e) After 24 h, cells were harvested and homogenized. Endolysosomal (heavy membrane, HM) and cytosol (supernatant, SUP) fractions of homogenate were collected and analyzed by immunoblotting. Lamp2 was used as a heavy membrane (HM) marker. The results were the representative from three different experiments. (f) After 12 h, cells were stained with anti-XIAP and anticleaved caspase-3 antibodies. Images were obtained by confocal microscopy. Scale bars, 10 μm. (g) After 24 h, viable cell numbers were counted after staining with trypan blue and indicated as a percentage of IL-2(+). Values were mean±S.D. from three different experiments. *P<0.005, **P<0.05
CTSB or CTSD were released from lysosomes to the cytosol.36, 37, 38 Lysosomal ceramide activates CTSD through its release from lysosome membranes to the cytosol.28 To examine CTSB release by IL-2(−) and the effect of desipramine on CTSB release, we separated cytosol and heavy membrane fractions, which contain lysosome membranes. Interestingly, IL-2(−) induced CTSB release from lysosome to cytosol, and its release was inhibited by desipramine treatment (Figure 5e). According to inhibition of CTSB, XIAP degradation, which elicits cell survival, was also suppressed by desipramine treatment (Figure 5g).
IL-2(−) induced caspase-dependent apoptosis. Thus we assessed caspase-3 activation during desipramine treatment. Interestingly, desipramine inhibited XIAP degradation but had no effect on caspase-3 activation (Figure 5e). However, XIAP colocalized with cleaved caspase-3 during desipramine treatment. Desipramine treatment inhibited XIAP downregulation and suppressed apoptosis (Figure 5g).
To confirm the effect of desipramine treatment, we established ASM-knockdown cells by using ASM-specific shRNA lentivirus. As shown in Figures 6a and b, ASM protein and activity were significantly reduced by ASM-knockdown cells (shASM) compared with control cells (shSCR). According to knockdown of ASM, XIAP degradation and PARP cleavage by IL-2(−) were blocked in shASM cells (Figure 6c). In addition, cell survival was also improved by ASM knockdown (Figure 6d). These results suggested that IL-2(−)-activated ASM increased lysosomal ceramide, which activates CTSB via its release from the lysosome to cytosol for XIAP degradation.

Knockdown of ASM inhibited IL-2(−)-mediated apoptosis. (a) Establishment of ASM knockdown KHYG-1 cells by using lentiviral particles of ASM-specific shRNA (shASM). Control cells (shSCR) were established as described in Figure 4e. ASM and actin were detected by western blotting analysis. (b) ASM activity was measured by using C6-NBD-SM as substrates. (c) After 24 h of treatment with IL−2(−) or IL-2(+), each protein was detected by immunoblotting. (d) After 36 h, viable cell numbers were counted after trypan blue staining and indicated as a percentage of IL-2(+) in each cell. Values are mean±S.D. from four different experiments. *P<0.005
Exogenous ceramide mimics IL-2(-) to activate CTSB and mediate XIAP degradation
From the above data, endogenous ceramide induced by IL-2(−) mediated CTSB activation, XIAP degradation, and caspase-dependent apoptosis. Previously, we have used permeable short chain N-acetyl sphingosine (C2-ceramide) to mimic the effects of endogenous ceramide on cell death, differentiation, and autophagy.39, 40, 41 Here, to examine whether exogenous C2-ceramide also induces ceramide-CTSB-mediated apoptosis, we treated KHYG-1 cells with C2-ceramide. As shown in Figure 7a, we measured ceramide amounts by liquid chromatography-electrospray ionization tandem mass spectrometry (LC-MS/MS) after treatment with C2-ceramide. Some ceramide species such as d18:1/16:0, d18:1/22:0, d18:1/24:1, and d18:1/24:0 were significantly elevated by C2-ceramide treatment and were same species induced by IL-2(−) treatment (Figures 2c and 7a). To elucidate whether exogenous ceramide also activates CTSB through its release from the lysosome to cytosol, we measured in vivo CTSB activity and examined the release of CTSB in C2-ceramide treatment (Figures 7b and c). After 12 h of treatment, C2-ceramide activated CTSB compared with vehicle treatment (control) (Figure 7b). Moreover, CTSB release from the lysosome to cytosol was also induced by C2-ceramide treatment (Figure 7c). Next, we assessed whether C2-ceramide-activated CTSB affected XIAP degradation. As shown in Figure 7d, C2-ceramide induced decrease of XIAP protein. Furthermore, CA-074 Me inhibited the degradation of XIAP induced by C2-ceramide (Figure 7d). These results suggested that exogenous ceramide also accelerates release and activation of CTSB with endogenous ceramide accumulation and mediates CTSB-dependent XIAP degradation.

C2-ceramide induced activation and release of CTSB. (a) Cells were treated with 50 μM C2-ceramide (C2-Cer) for 6 h. Ceramide amounts were measured by LC-MS/MS. Values are mean±S.D. from three different experiments. *P<0.005. (b) Cells were treated with 50 μM C2-ceramide for 12 h. CTSB activity was measured by the Magic Red CTSB Activity Kit. Images were obtained by fluorescent microscopy. Values are mean±S.D. from three different experiments. *P<0.005, **P<0.05. (c) Release of CTSB was assessed by western blotting analysis after separating fractions of heavy membrane (HM) and cytosol (SUP). (d) XIAP levels were measured by immunoblotting using anti-XIAP antibodies. Results are representative of three independent experiments
Exogenous ceramide induced XIAP degradation and caspase-dependent apoptosis
Finally, we examined the effect of exogenous ceramide on XIAP degradation and caspase-dependent apoptosis. C2-ceramide treatment reduced cytosolic XIAP protein by western blotting analysis and immunocytochemistry (Figures 8a and b). Active caspase-3 was detected by C2-ceramide treatment as demonstrated by XIAP degradation (Figure 8b). Moreover, C2-ceramide induced in vivo caspase-3 activation and apoptosis (Figures 8c and d). These data suggested that C2-ceramide mimicked IL-2(−)-induced apoptosis by XIAP degradation and caspase-3 activation.

C2-ceramide induced apoptosis with XIAP degradation. (a) KHYG-1 cells were treated with 50 μM C2-ceramide (C2-Cer) for the indicated times. XIAP protein was detected by western blotting analysis. (b) After 12 h of C2-Cer treatment, cells were stained with anti-XIAP and anticleaved caspase-3 antibodies. Nuclei were counterstained with DAPI. Images were obtained by confocal microscopy. Scale bars, 10 μm. (c) In vivo caspase-3/-7 activity was measured by fluorescent substrate of caspase-3/7 (FAM-DEVD-FMK). Images were obtained by fluorescent microscopy. Values are mean±S.D. from three different experiments. *P<0.005. (d) After 12 h, viable cell numbers were counted after staining with trypan blue and indicated as a percentage of vehicle treatment. Values were mean±S.D. from three different experiments. *P<0.005
Discussion
KHYG-1 NK/T lymphoma cells require IL-2 for their proliferation and survival and undergo apoptosis by IL-2(−) even in the presence of serum. Thus it is important to understand the mechanism of IL-2(−)-induced apoptosis to develop novel treatments for drug-resistant NK/T cell lymphoma. We first showed that IL-2(−) induced caspase-dependent apoptosis, characterized by PS externalization and cleavage of caspase-8, -9, and -3, and the addition of IL-2 rescued cell death. Interestingly, although proteinase activity of caspase-3 was inhibited by IL-2 rescue, the 17–19-kD cleaved form of caspase-3 was observed. Furthermore, cleaved caspase-3 p17 did not traffic into the nucleus after IL-2 rescue while IL-2(−)-induced active caspase-3 accumulated in the nucleus to mediate the degradation of PARP. The precise mechanism of nuclear transport of caspase-3 has been clarified, but the translocation of caspase-3 p17 with more processing form p12 into the nucleus might be involved in Fas-induced apoptosis. Previous studies have demonstrated that p3-recognition site of caspase-3 is important for its nuclear entry.42, 43 Indeed, our data showed that more short form of cleaved caspase-3 (12 kD) was slightly but reliably detected in only IL-2(−), suggesting that caspase-3 was not cleaved completely in IL-2 rescue. In addition, caspase-3 p17 remained in the cytosol and did not move into the nucleus after rescue of apoptosis by IL-2 supplementation. These results suggested that the mechanism that inhibits the entry of caspase-3 p17 into the nucleus exists in the cytosol.
It is well known that ASM localizes in lysosomes. During IL-2(−) conditions, ASM-induced increase in ceramide was detected in lysosomes using anti-ceramide antibodies that colocalized with a lysosome marker Lamp1. After IL-2 rescue, the ceramide content in lysosomes decreased to the IL-2(+) control level. The molecular species of IL-2(−)-increased ceramide are of d18:1/16:0 and d18:1/24:1 types. We can detect that the increase of molecular species of ceramide by a diverse of stressors appears to be similar in pattern regardless of the mechanisms of PCD (data not shown). The localization of ceramide generated by the different pathways may be more critical for the regulation of cell death by ceramide signal, but at present evidence is ambiguous.
We detected in vitro activities of caspases in lysates of IL-2(−) and IL-2 rescued cells (data not shown). However, the in vivo activity of caspase-3 was significantly suppressed by IL-2 rescue. Because XIAP can directly bind to caspase-3 to inhibit cleaved activation,14 we focused on the regulation of XIAP protein, a member of the IAP family, by ASM-induced lysosomal ceramide.14, 16 A decrease of XIAP by IL-2(−) was restored by IL-2 rescue. This reduction was due to protein degradation but not the suppression of mRNA expression in XIAP (data not shown). It was reported that XIAP expression was controlled by activated NF-κB44 and that the PI3-K/Akt pathway, upstream of NF-κB, regulated XIAP.45 We also detected NF-κB activation and enhanced XIAP mRNA expression by IL-2 rescue (data not shown). This suggests that the increase of XIAP in cell proliferation is regulated transcriptionally, whereas the decrease of XIAP induced by IL-2(−) is due to degradation at the protein level.
Why could not XIAP inhibit caspase cleavage? Recently, some reports showed that cleaved caspase has other effects such as inflammatory activation without apoptosis. Kavanagh et al.46 demonstrated that cytoplasmic cleaved caspase-3 induces pro-inflammatory activation through PKCδ but not apoptosis in microglia. Pro-inflammatory response activates NF-kB pathway. Moreover, Cheng et al.47 showed that ionizing radiation activates PKCδ through caspase-3/7 and leads production of growth factors via Akt pathway in pancreatic cancer cells. Indeed, rescue activated NF-κB pathway and cell proliferation (data not shown). Thus cytoplasmic cleaved caspase might be related in cell proliferation but not apoptosis in rescue condition.
Recently, it was reported that the ceramide analogue LCL85 enhanced Fas-induced apoptosis by inhibiting XIAP.48 We here demonstrated that regulation of ceramide in lysosomes has a role in the degradation of XIAP. What is the mechanism by which XIAP is degraded by lysosomal ceramide? Mechanisms that decrease XIAP protein include degradation by ubiquitination and proteases and/or transcriptional regulation.49 Ceramide mediates apoptosis, and many molecules, including ceramide-activated protein phosphatases and protein kinase C ζ, are involved in its signaling.50, 51 Especially, lysosomal aspartic protease CTSD is reported to be activated by ASM-generated ceramide to induce apoptotic cell death in response to TNF signaling.28, 35 Lysosomal ceramide generated by ASM bound to CTSD mediated its translocation to the cytosol, following activation of the mitochondrial apoptotic pathway. 28 Interestingly, we found that an inhibitor of cysteine protease CTSB (CA-074 Me), but not CTSD (pepstatin A), inhibited XIAP degradation, and activation of CTSB was detected after IL-2(−) treatment. Inhibitors of CTSB suppressed IL-2(−)-mediated apoptotic cell death, suggesting a role for CTSB, but not CTSD, in apoptosis. However, previous studies indicated a role for CTSB in tumor progression.52 The increased expression of CTSB was related to the invasion and proliferation of melanoma cells,9 and CTSB decreased dramatically after serum-deprivation-induced apoptosis, whereas CTSD increased, suggesting a balance between CTSB and CTSD.53 In the current study, we showed that lysosomal ceramide-activated CTSB induced caspase activation through XIAP degradation because an inhibitor of CTSB CA-074 Me was effective in blocking the apoptotic pathway. However, we could not detect the cleavage site of CTSB in XIAP protein or direct digestion of XIAP using cell lysates and recombinant CTSB (data not shown). Bien et al.54 showed that doxorubicin activated CTSB and CTSB-mediated caspase-dependent apoptosis in HeLa cells. In addition, the downregulation of XIAP was also induced by doxorubicin and was inhibited by CTSB siRNA or CA-074 Me treatment. These data support our finding that CTSB regulates XIAP degradation.
CA-074 Me did not inhibit the increase of ceramide in lysosomes during IL-2(−), suggesting that ASM-induced ceramide generation is upstream of CTSB activation. Although CTSB has not been reported to bind to ceramide, previous studies showed that CTSB release is induced by alterations of lysosomal membrane permeabilization (LMP) in response to various stimuli, which activates caspase-mediated apoptosis.55 Thus lysosomal ceramide might induce LMP to release CTSB to the cytosol, but the precise mechanism of CTSB activation by lysosomal ceramide is unknown.
ASM activation and ceramide generation have been reported in various signaling pathways triggered by numerous stimuli,25, 56 chemotherapeutic drugs,30, 57 irradiation,58, 59 or pathogens.27, 60 In the present study, we showed IL-2(−) activated ASM, but not NSM, and induced ceramide accumulation in lysosomes, suggesting that lysosomal ceramide is critical for IL-2(−)-induced apoptosis. Indeed, an inhibitor of ASM, desipramine, inhibited IL-2(−)-induced apoptosis by suppressing ceramide generation in lysosomes. Desipramine also inhibited the nuclear activation of caspase-3 through the degradation of XIAP by CTSB activation.
Finally, we investigated whether exogenous C2-ceramide mimics IL-2(−)-mediated CTSB activation, XIAP degradation, and caspase-dependent NK/T lymphoma cell apoptosis. C2-ceramide increased apoptosis by generating physiological d18:1/16:0 and d18:1/24:1 ceramides similar to IL-2 deprivation. The increase of ceramides induced activation of CTSB and subsequent XIAP degradation. Huang et al.61 described that exogenous treatment with C2-ceramide triggered lysosomal pathways in T cell hybridomas and A549 lung adenocarcinoma cells and that C2-ceramide might have no direct effect on lysosomal function because the inhibition of ASM did not affect LMP or apoptosis. Recently, sphingosine, a metabolite of ceramide via ceramidase, was reported to mediate LMP and relocation of CTSB in TNF-α-treated hepatoma cells.55 Thus sphingosine generated by acid ceramidase from ASM-generated ceramide might activate CTSB during IL-2(−). In fact, IL-2(−) also increased sphingosine (18 : 1) (from 3.35±0.85 pmol/1 × 106 cells in IL-2(+) to 4.97±0.27 pmol/1 × 106 cells in IL-2(−)) (data not shown). However, amount of ceramide (459.3±21.4 pmol/1 × 106 cells in IL-2(−)) was more than 100-fold of sphingosine. In addition, we investigated the effects of inhibitors of enzymes related in ceramide metabolism on C2-ceramide-induced cell death (Supplementary Figure S1). At first, the treatment of desipramine with C2-ceramide had no effect on cell survival. This result suggested that ASM is not involved in cellular metabolism of C2-ceramide in our case. Next, we used inhibitors of ceramide synthase and acid ceramidase, which inhibit the metabolism of ceramide from sphingosine and of sphingosine from ceramide, respectively. Fumonisin B1, which is ceramide synthase inhibitor to block ceramide production from sphingosine, recovered cell survival compared with C2-ceramide only. Inversely, acid ceramidase inhibitor D-NMAPPD enhanced C2-ceramide-medaited cell death. Acid ceramidase produces sphingosine from ceramide in lysosome. These results demonstrated that ceramide but not sphingosine is related in C2-ceramide-induced apoptosis. In addition, some reports that acid ceramidase is implicated in cell survival of malignant cells supported our data that acid ceramidase has inhibitory effect on C2-ceramide-mediated apoptosis.62, 63, 64 Thus we believe ASM-generated ceramide affected IL-2(−)-induced apoptosis.
In summary, our present work demonstrated a novel pathway related to lysosomal ceramide, CTSB, and XIAP in IL-2(−)-induced NK/T lymphoma cell apoptosis. Lysosomal ceramide generated by ASM induced the release of CTSB into the cytosol and the degradation of XIAP, resulting in the nuclear entry of active caspase-3 and subsequent apoptosis. The pathophysiological implication of this lysosomal ceramide/CTSB/XIAP axis in apoptotic cell death should be clarified in the future to develop a targeting therapy for NK/T lymphoma.
Materials and Methods
Materials
Materials were purchased as follows: human recombinant IL-2 (Imunase 35) (Shionogi Pharmaceutical, Osaka, Japan); C6-NBD ceramide, C6-NBD sphingomyelin, and C2-ceramide (Matreya, Pleasant Gap, PA, USA); CTSB specific inhibitor IV (CA-074 Me) (Merck Millipore, Darmstadt, Germany); pepstatin A and desipramine (Sigma-Aldrich, St. Louis, MO, USA); anti-caspase-8, anti-caspase-9, anti-PARP, antilysosomal-associated membrane protein 1 (Lamp1), anti-Lamp2, anti-ASM, anti-CTSB, anti-CTSD, and anti-β-actin antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA); Annexin V-conjugated FITC and monoclonal antibody against XIAP (BD Biosciences, Beverly, MA, USA); antiactive caspase-3 (p17) antibody and horseradish peroxidase–conjugated secondary antibodies (Promega, Madison, WI, USA); anti-caspase-3, anti-pan-cadherin, and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies (Cell signaling, Danvers, MA, USA); and anti-ceramide monoclonal IgM Ab clone (NHCER-2) was produced previously in our lab.65 Alexa Fluor-conjugated (AF-conjugated) secondary antibodies were from Molecular Probes (Eugene, OR, USA). 4’,6-Diamidino-2-phenylindole (DAPI) was obtained from Roche Applied Science (Penzberg, Germany).
Cell culture and counting of viable cells
IL-2 deprivation (IL-2(−)) and re-supplementation of human KHYG-1 cells were described previously (Figure 1a).(3) Cells were grown in RPMI 1640 (Sigma) supplemented with 10% fetal bovine serum and IL-2 (100 mU/ml) (IL-2–containing medium, IL-2(+)) at 37 °C in a humidified atmosphere containing 5% CO2. For IL-2 rescue experiments, cells were cultured in IL-2-depleted medium for 12 hours. Then, IL-2 (100 mU/ml) was supplied to media, and cells were kept thereafter (rescue). IL-2(−) indicated that cells were cultured in IL-2-deprived medium. Viable cell numbers were counted by 0.25% (w/v) trypan blue dye exclusion method at the indicated period.
For establishments of ASM or CTSB knockdown cells, lentiviral particles of shRNA were obtained from Santa Cruz. Cells were infected with control shRNA (shSCR, sc-108080), ASM-specific shRNA (shASM, sc-41650-v), and CTSB-specific shRNA (shCTSB, sc-29238-v) lentiviral particles after the treatment with 1 μg/ml polybrene. Then cells were treated with 2 μg/ml puromycin for establishment of knockdown cell lines. Efficiency of knockdown was checked by western blotting analysis.
Detection of apoptotic cells by flow cytometry
Apoptotic cells were detected by PS exposure by staining with Annexin V-FITC and analyzed by flow cytometry. Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and suspended in Annexin V binding buffer (BD Biosciences). After 15 min of staining with Annexin V–FITC and 7AAD, apoptotic cells were analyzed by flow cytometry (Gallios, Beckman Coulter, Miami, FL, USA) according to the manufacturer’s protocol. For each group, a minimum of 10 000 cells were collected.
Analysis of in vivo caspase and CTSB activities
In vivo caspase-3/7 activity was measured by FAM-fluorochrome inhibitor of caspases (FLICA) Caspase 3&7 Assay Kit (Immunochemistry Technologies, Bloomington, MN, USA) according to the manufacturer’s information. Briefly, cells were incubated with the FAM-DEVD-fluoromethylketone (FMK) reagent for 1 h at 37 °C. After washing with PBS, cells were fixed with 0.1% (w/v) formaldehyde solution. In vivo CTSB activity was performed using Magic Red CTSB Activity Kit (Immunochemistry Technologies) according to the manufacturer’s protocol. Cells were incubated with Magic Red CTSB Substrate (Immunochemistry Technologies) for 1 h at 37 °C. The cells were pelleted after centrifugation, washed with PBS, and fixed with 0.1% formaldehyde. Fluorescence was measured by a fluorescent microplate reader Infinite 500 (Tecan Group Ltd., Männedorf, Switzerland), or at least 200 cells were counted and indicated as the percentage of positive cells per total cells. Images were obtained by fluorescent microscopy Leica DMRB or confocal microscopy Leica TSC SP2 (Leica Microsystems, Wetzlar, Germany).
Western blotting analysis
KHYG-1 cells were harvested, washed with PBS, and lysed in lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1% (w/v) Triton X-100, 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). After incubation on ice for 20 min, debris was removed by centrifugation at 2000 × g for 10 min at 4 °C. Supernatant was used as a loading sample. Proteins (30 μg) were subjected to SDS-poly-acrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Nonspecific binding was blocked by incubation of the membrane with PBS containing 0.1% (w/v) Tween-20 (PBS-T) and 5% (w/v) non-fat dried milk for 20 min at room temperature. Then membrane was incubated with primary antibodies overnight at 4 °C and with secondary antibodies for 45 min at room temperature. Immunoreactive protein bands were visualized using an ECL-peroxidase detection system (Amersham Biosciences, Piscataway, NJ, USA) and LAS-4000 (Fujifilm, Tokyo, Japan).
Acid and NSM activities
KHYG-1 cells were sonicated in lysis buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.1% (w/v) Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2.5 μg/ml leupeptin, and 2.5 μg/ml aprotinin) and incubated on ice for 30 minutes. The homogenate was centrifuged at 10 000 × g for 10 min at 4 °C, and the supernatant was used as an enzyme source. Proteins (100 μg) were mixed in reaction buffer for magnesium-dependent NSM (0.1 M Tris-HCl, pH 7.5, 10 μM C6-NBD sphingomyelin, 10 mM MgCl2, 0.1% (w/v) Triton X-100, 5 mM dithiothreitol) or for ASM (0.1 M sodium acetate (pH 5.0), 10 μM C6-NBD sphingomyelin, 0.1% (w/v) Triton X-100) and incubated for 90 min at 37 °C. Lipids were extracted using chroloform/methanol, applied to thin layer chromatography plates, and separated in chloroform/methanol/12 mM MgCl2 in D.W. (65 : 25 : 4, v/v/v). Fluorescent lipids were detected using LAS-4000 (Fujifilm) and quantified using MultiGauge 3.1 (Fujifilm).
Ceramide measurement by LC-MS/MS
Lipid extraction from KHYG-1 cells (1 × 106 cells) and measurements of ceramide species in lipid extracts were performed using LC-MS/MS as described previously.66 The amounts of ceramide species with various carbon chains (d18:1/16:0, d18:0/16:0, d18:1/18:1, d18:1/18:0, d18:1/20:1, d18:1/20:0, d18:1/22:1, d18:1/22:0, d18:1/24:1, d18:1/24:0, d18:1/26:0, d16:1/18:1, d16:1/20:0, and d18:0/24:1) were determined.
Immunocytochemistry
For detection of active caspase-3 and XIAP, cells were treated by the indicated conditions, washed with ice-cold PBS, cytospun onto slides, and then fixed with 2% formaldehyde for 15 min at 4 °C. Fixed cells were permeabilized with PBS containing 0.1% Triton X-100 for 5 min and incubated with PBS containing 2% (w/v) BSA for 30 min at room temperature. Cells were washed with PBS and then incubated with primary antibodies for 90 min at room temperature. After washing with PBS, AF488- or AF546-conjugated anti-IgG antibodies were incubated for 45 min. For determination of lysosomal ceramide, fixed cells were treated with anti-ceramide antibody and anti-Lamp1 antibody for 90 min at room temperature. Then AF488-conjugated anti-IgM and AF546-conjugated anti-IgG antibodies were incubated for 45 min at room temperature. Nuclei were counterstained with DAPI.
Cell fractionation
Lysosome fraction was collected by using the Lysosome Enrichment Kit for Tissue and Cultured Cells according to the manufacturer’s protocol (Thermo Scientific, 89839, Rockford, IL, USA). Cells (1 × 107 cells) were treated with IL-2(+) or IL-2(−) for 24 h, and cell pellets were used for lysosome isolation. Whole cell lysate before lysosome collection and isolated lysosome fractions were then subjected to western blotting analysis to check the purity of the isolated lysosome and measurement of SM by LC-MS/MS. Anti-Lamp2 (lysosome), anti-pan-cadherin (plasma membrane), and anti-GAPDH (cytosol) antibodies were used as markers of each organelle.
CTSB and CTSD release
The release of CTSB and CTSD from the endolysosomal compartment was analyzed by heavy membrane fractionation.67 Cells were lysed in homogenization buffer (10 mM Hepes-KOH, pH 7.4, 1 mM EDTA, 0.25 M sucrose, 1 mM PMSF, inhibitor cocktail (Roche)), incubated on ice for 40 min, and homogenized by 27-G syringes. Homogenates were centrifuged at 300 × g for 5 min, and unbroken cells were removed. Then lysate was centrifuged at 20 000 × g for 30 min, and supernatants contained the cytoplasmic fraction. Pellets were resolved in homogenization buffer containing 1% Triton X-100 and used for heavy membrane fraction, including lysosomes. Proteins (40 μg) were used for western blotting to detect the release of CTSB and CTSD from the heavy membrane fraction to the cytosol fraction. Anti-Lamp2 antibody was used as a lysosomal marker.
Statistical analysis
Comparisons between two groups were carried out using the unpaired Student’s t-test.
Abbreviations
- ASM:
-
acid sphingomyelinase
- CTSB:
-
cathepsin B
- CTSD:
-
cathepsin D
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- IL-2:
-
interleukin-2
- LC-MS/MS:
-
liquid chromatography-electrospray ionization tandem mass spectrometry
- NK/T:
-
natural killer T
- NSM:
-
neutral sphingomyelinase
- PS:
-
phosphatidylserine
- shRNA:
-
short hairpin RNA
- SMS:
-
sphingomyelin synthase
- XIAP:
-
X-linked inhibitor of apoptosis protein
References
Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia 2000; 14: 922–930.
Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol 2005; 33: 1160–1171.
Taguchi Y, Kondo T, Watanabe M, Miyaji M, Umehara H, Kozutsumi Y et al. Interleukin-2-induced survival of natural killer (NK) cells involving phosphatidylinositol-3 kinase-dependent reduction of ceramide through acid sphingomyelinase, sphingomyelin synthase, and glucosylceramide synthase. Blood 2004; 104: 3285–3293.
Fiandalo MV, Kyprianou N . Caspase control: protagonists of cancer cell apoptosis. Exp Oncol 2012; 34: 165–175.
Repnik U, Stoka V, Turk V, Turk B . Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta 2012; 1824: 22–33.
Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem 2008; 283: 19140–19150.
Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem 2003; 278: 31401–31411.
Gogineni VR, Gupta R, Nalla AK, Velpula KK, Rao JS . uPAR and cathepsin B shRNA impedes TGF-beta1-driven proliferation and invasion of meningioma cells in a XIAP-dependent pathway. Cell Death Dis 2012; 3: e439.
Yin M, Soikkeli J, Jahkola T, Virolainen S, Saksela O, Holtta E . TGF-beta signaling, activated stromal fibroblasts, and cysteine cathepsins B and L drive the invasive growth of human melanoma cells. Am J Pathol 2012; 181: 2202–2216.
Morchang A, Panaampon J, Suttitheptumrong A, Yasamut U, Noisakran S, Yenchitsomanus PT et al. Role of cathepsin B in dengue virus-mediated apoptosis. Biochem Biophys Res Commun 2013; 438: 20–25.
Paulsen M, Ussat S, Jakob M, Scherer G, Lepenies I, Schutze S et al. Interaction with XIAP prevents full caspase-3/-7 activation in proliferating human T lymphocytes. Eur J Immunol 2008; 38: 1979–1987.
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410: 112–116.
Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H . Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 2001; 104: 781–790.
Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001; 104: 791–800.
Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS . XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J 2005; 24: 645–655.
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 2003; 11: 519–527.
Inoue H, Shiraki K, Murata K, Sugimoto K, Kawakita T, Yamaguchi Y et al. Adenoviral-mediated transfer of p53 gene enhances TRAIL-induced apoptosis in human hepatocellular carcinoma cells. Int J Mol Med 2004; 14: 271–275.
Marienfeld C, Yamagiwa Y, Ueno Y, Chiasson V, Brooks L, Meng F et al. Translational regulation of XIAP expression and cell survival during hypoxia in human cholangiocarcinoma. Gastroenterology 2004; 127: 1787–1797.
Friesen C, Roscher M, Hormann I, Leib O, Marx S, Moreno J et al. Anti-CD33-antibodies labelled with the alpha-emitter Bismuth-213 kill CD33-positive acute myeloid leukaemia cells specifically by activation of caspases and break radio- and chemoresistance by inhibition of the anti-apoptotic proteins X-linked inhibitor of apoptosis protein and B-cell lymphoma-extra large. Eur J Cancer 2013; 49: 2542–2554.
Ibrahim AM, Mansour IM, Wilson MM, Mokhtar DA, Helal AM, Al Wakeel HM . Study of survivin and X-linked inhibitor of apoptosis protein (XIAP) genes in acute myeloid leukemia (AML). Lab Hematol 2012; 18: 1–10.
Lin H, Chen C, Li X, Chen BD . Activation of the MEK/MAPK pathway is involved in bryostatin1-induced monocytic differenciation and up-regulation of X-linked inhibitor of apoptosis protein. Exp Cell Res 2002; 272: 192–198.
Hannun YA, Obeid LM . Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008; 9: 139–150.
Morad SA, Cabot MC . Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer 2013; 13: 51–65.
Young MM, Kester M, Wang HG . Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res 2013; 54: 5–19.
Dumitru CA, Gulbins E . TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 2006; 25: 5612–5625.
Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K et al. CD95 signaling via ceramide-rich membrane rafts. J Biol Chem 2001; 276: 20589–20596.
Grassme H, Cremesti A, Kolesnick R, Gulbins E . Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 2003; 22: 5457–5470.
Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 2004; 11: 550–563.
Edelmann B, Bertsch U, Tchikov V, Winoto-Morbach S, Perrotta C, Jakob M et al. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J 2011; 30: 379–394.
Dumitru CA, Sandalcioglu IE, Wagner M, Weller M, Gulbins E . Lysosomal ceramide mediates gemcitabine-induced death of glioma cells. J Mol Med (Berl) 2009; 87: 1123–1132.
Chen H, Hao Y, Wang L, Jia D, Ruan Y, Gu J . Sodium arsenite down-regulates the expression of X-linked inhibitor of apoptosis protein via translational and post-translational mechanisms in hepatocellular carcinoma. Biochem Biophys Res Commun 2012; 422: 721–726.
Yoon JY, Szwajcer D, Ishdorj G, Benjaminson P, Xiao W, Kumar R et al. Synergistic apoptotic response between valproic acid and fludarabine in chronic lymphocytic leukaemia (CLL) cells involves the lysosomal protease cathepsin B. Blood Cancer J 2013; 3: e153.
Jenkins RW, Canals D, Hannun YA . Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal 2009; 21: 836–846.
Cesen MH, Pegan K, Spes A, Turk B . Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res 2012; 318: 1245–1251.
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R et al. Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J 1999; 18: 5252–5263.
Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 2010; 17: 1167–1178.
Cheriyath V, Kuhns MA, Kalaycio ME, Borden EC . Potentiation of apoptosis by histone deacetylase inhibitors and doxorubicin combination: cytoplasmic cathepsin B as a mediator of apoptosis in multiple myeloma. Br J Cancer 2011; 104: 957–967.
Granato M, Lacconi V, Peddis M, Lotti LV, Renzo LD, Gonnella R et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis 2013; 4: e730.
Okazaki T, Bielawska A, Bell RM, Hannun YA . Role of ceramide as a lipid mediator of 1 alpha,25-dihydroxyvitamin D3-induced HL-60 cell differentiation. J Biol Chem 1990; 265: 15823–15831.
Iwai K, Kondo T, Watanabe M, Yabu T, Kitano T, Taguchi Y et al. Ceramide increases oxidative damage due to inhibition of catalase by caspase-3-dependent proteolysis in HL-60 cell apoptosis. J Biol Chem 2003; 278: 9813–9822.
Taniguchi M, Kitatani K, Kondo T, Hashimoto-Nishimura M, Asano S, Hayashi A et al. Regulation of autophagy and its associated cell death by "sphingolipid rheostat": reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem 2012; 287: 39898–39910.
Kamada S, Kikkawa U, Tsujimoto Y, Hunter T . Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J Biol Chem 2005; 280: 857–860.
Luo M, Lu Z, Sun H, Yuan K, Zhang Q, Meng S et al. Nuclear entry of active caspase-3 is facilitated by its p3-recognition-based specific cleavage activity. Cell Res 2010; 20: 211–222.
Qin Y, Auh S, Blokh L, Long C, Gagnon I, Hamann KJ . TNF-alpha induces transient resistance to Fas-induced apoptosis in eosinophilic acute myeloid leukemia cells. Cell Mol Immunol 2007; 4: 43–52.
Gagnon V, Van Themsche C, Turner S, Leblanc V, Asselin E . Akt and XIAP regulate the sensitivity of human uterine cancer cells to cisplatin, doxorubicin and taxol. Apoptosis 2008; 13: 259–271.
Kavanagh E, Rodhe J, Burguillos MA, Venero JL, Joseph B . Regulation of caspase-3 processing by cIAP2 controls the switch between pro-inflammatory activation and cell death in microglia. Cell Death Dis 2014; 5: e1565.
Cheng J, Tian L, Ma J, Gong Y, Zhang Z, Chen Z et al. Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase Cdelta activation in pancreatic ductal adenocarcinoma. Mol Oncol 2015; 9: 105–114.
Paschall AV, Zimmerman MA, Torres CM, Yang D, Chen MR, Li X et al. Ceramide targets xIAP and cIAP1 to sensitize metastatic colon and breast cancer cells to apoptosis induction to suppress tumor progression. BMC Cancer 2014; 14: 24.
Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD . Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000; 288: 874–877.
Futerman AH, Riezman H . The ins and outs of sphingolipid synthesis. Trends Cell Biol 2005; 15: 312–318.
Chalfant CE, Ogretmen B, Galadari S, Kroesen BJ, Pettus BJ, Hannun YA . FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem 2001; 276: 44848–44855.
Yan S, Sameni M, Sloane BF . Cathespin B and human tumor progression. Biol Chem 1998; 379: 113–123.
Shibata M, Kanamori S, Isahara K, Ohsawa Y, Konishi A, Kametaka S et al. Participation of cathepsins B and D in apoptosis of PC12 cells following serum deprivation. Biochem Biophys Res Commun 1998; 251: 199–203.
Bien S, Rimmbach C, Neumann H, Niessen J, Reimer E, Ritter CA et al. Doxorubicin-induced cell death requires cathepsin B in HeLa cells. Biochem Pharmacol 2010; 80: 1466–1477.
Ullio C, Casas J, Brunk UT, Sala G, Fabrias G, Ghidoni R et al. Sphingosine mediates TNFalpha-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J Lipid Res 2012; 53: 1134–1143.
Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z et al. Ceramide enables fas to cap and kill. J Biol Chem 2001; 276: 23954–23961.
Lacour S, Hammann A, Grazide S, Lagadic-Gossmann D, Athias A, Sergent O et al. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res 2004; 64: 3593–3598.
Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003; 300: 1155–1159.
Charruyer A, Grazide S, Bezombes C, Muller S, Laurent G, Jaffrezou JP . UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J Biol Chem 2005; 280: 19196–19204.
Jan JT, Chatterjee S, Griffin DE . Sindbis virus entry into cells triggers apoptosis by activating sphingomyelinase, leading to the release of ceramide. J Virol 2000; 74: 6425–6432.
Huang WC, Chen CL, Lin YS, Lin CF . Apoptotic sphingolipid ceramide in cancer therapy. J Lipids 2011; 2011: 565316.
Beckham TH, Cheng JC, Lu P, Marrison ST, Norris JS, Liu X . Acid ceramidase promotes nuclear export of PTEN through sphingosine 1-phosphate mediated Akt signaling. PLoS One 2013; 8: e76593.
Beckham TH, Cheng JC, Lu P, Shao Y, Troyer D, Lance R et al. Acid ceramidase induces sphingosine kinase 1/S1P receptor 2-mediated activation of oncogenic Akt signaling. Oncogenesis 2013; 2: e49.
Camacho L, Meca-Cortes O, Abad JL, Garcia S, Rubio N, Diaz A et al. Acid ceramidase as a therapeutic target in metastatic prostate cancer. J Lipid Res 2013; 54: 1207–1220.
Kawase M, Watanabe M, Kondo T, Yabu T, Taguchi Y, Umehara H et al. Increase of ceramide in adriamycin-induced HL-60 cell apoptosis: detection by a novel anti-ceramide antibody. Biochim Biophys Acta 2002; 1584: 104–114.
Ogiso H, Taniguchi M, Araya S, Aoki S, Wardhani LO, Yamashita Y et al. Comparative analysis of biological sphingolipids with glycerophospholipids and diacylglycerol by LC-MS/MS. Metabolites 2014; 4: 17.
Ha SD, Ham B, Mogridge J, Saftig P, Lin S, Kim SO . Cathepsin B-mediated autophagy flux facilitates the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm. J Biol Chem 2010; 285: 2120–2129.
Acknowledgements
We are grateful to Dr. Mayumi Hashimoto-Nishimura (Tottori University) and Dr. Satoshi Asano (Hiroshima University) for critical discussions. This study was supported by: the Takeda Science Foundation (2012); a joint research project with Shalome Co., Ltd. (2012–2015); ONO Pharmaceutical Co., Ltd. (2012–2014); the JSPS KAKENHI (Grant Number 25870853 (to MT) and 21890144 (to KK)); a grant from Strategic Research Foundation Grant-aided Project for Private Universities from the MEXT (No. S1201004; H2012-16); SENSHIN Medical Research Foundation; Mizutani Foundation for Glycoscience (2013–14); and the Collaborative Research from Kanazawa Medical University (C2012-4, C2013-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Chipuk
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Taniguchi, M., Ogiso, H., Takeuchi, T. et al. Lysosomal ceramide generated by acid sphingomyelinase triggers cytosolic cathepsin B-mediated degradation of X-linked inhibitor of apoptosis protein in natural killer/T lymphoma cell apoptosis. Cell Death Dis 6, e1717 (2015). https://doi.org/10.1038/cddis.2015.82
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.82
This article is cited by
-
Endosomes, lysosomes, and the role of endosomal and lysosomal biogenesis in cancer development
Molecular Biology Reports (2020)
-
Targeting bone morphogenetic protein receptor 2 sensitizes lung cancer cells to TRAIL by increasing cytosolic Smac/DIABLO and the downregulation of X-linked inhibitor of apoptosis protein
Cell Communication and Signaling (2019)
-
Sphingolipids and their metabolism in physiology and disease
Nature Reviews Molecular Cell Biology (2018)
-
Acid sphingomyelinase mediates human CD4+ T-cell signaling: potential roles in T-cell responses and diseases
Cell Death & Disease (2017)
-
Ceramide mediates FasL-induced caspase 8 activation in colon carcinoma cells to enhance FasL-induced cytotoxicity by tumor-specific cytotoxic T lymphocytes
Scientific Reports (2016)

{kind=link}