
Abstract
Neuroinflammation is a striking hallmark of amyotrophic lateral sclerosis (ALS) and other neurodegenerative disorders. Previous studies have shown the contribution of glial cells such as astrocytes in TDP-43-linked ALS. However, the role of microglia in TDP-43-mediated motor neuron degeneration remains poorly understood. In this study, we show that depletion of TDP-43 in microglia, but not in astrocytes, strikingly upregulates cyclooxygenase-2 (COX-2) expression and prostaglandin E2 (PGE2) production through the activation of MAPK/ERK signaling and initiates neurotoxicity. Moreover, we find that administration of celecoxib, a specific COX-2 inhibitor, greatly diminishes the neurotoxicity triggered by TDP-43-depleted microglia. Taken together, our results reveal a previously unrecognized non-cell-autonomous mechanism in TDP-43-mediated neurodegeneration, identifying COX-2-PGE2 as the molecular events of microglia- but not astrocyte-initiated neurotoxicity and identifying celecoxib as a novel potential therapy for TDP-43-linked ALS and possibly other types of ALS.
Similar content being viewed by others


Main
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by the degeneration of motor neurons in the brain and spinal cord.1 Most cases of ALS are sporadic, but 10% are familial. Familial ALS cases are associated with mutations in genes such as Cu/Zn superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TARDBP) and, most recently discovered, C9orf72. Currently, most available information obtained from ALS research is based on the study of SOD1, but new studies focusing on TARDBP and C9orf72 have come to the forefront of ALS research.1, 2 The discovery of the central role of the protein TDP-43, encoded by TARDBP, in ALS was a breakthrough in ALS research.3, 4, 5 Although pathogenic mutations of TDP-43 are genetically rare, abnormal TDP-43 function is thought to be associated with the majority of ALS cases.1 TDP-43 was identified as a key component of the ubiquitin-positive inclusions in most ALS patients and also in other neurodegenerative diseases such as frontotemporal lobar degeneration,6, 7 Alzheimer’s disease (AD)8, 9 and Parkinson’s disease (PD).10, 11 TDP-43 is a multifunctional RNA binding protein, and loss-of-function of TDP-43 has been increasingly recognized as a key contributor in TDP-43-mediated pathogenesis.5, 12, 13, 14
Neuroinflammation, a striking and common hallmark involved in many neurodegenerative diseases, including ALS, is characterized by extensive activation of glial cells including microglia, astrocytes and oligodendrocytes.15, 16 Although numerous studies have focused on the intrinsic properties of motor neurons in ALS, a large amount of evidence showed that glial cells, such as astrocytes and microglia, could have critical roles in SOD1-mediated motor neuron degeneration and ALS progression,17, 18, 19, 20, 21, 22 indicating the importance of non-cell-autonomous toxicity in SOD1-mediated ALS pathogenesis.
Very interestingly, a vital insight of neuroinflammation research in ALS was generated by the evidence that both the mRNA and protein levels of the pro-inflammatory enzyme cyclooxygenase-2 (COX-2) are upregulated in both transgenic mouse models and in human postmortem brain and spinal cord.23, 24, 25, 26, 27, 28, 29 The role of COX-2 neurotoxicity in ALS and other neurodegenerative disorders has been well explored.30, 31, 32 One of the key downstream products of COX-2, prostaglandin E2 (PGE2), can directly mediate COX-2 neurotoxicity both in vitro and in vivo.33, 34, 35, 36, 37 The levels of COX-2 expression and PGE2 production are controlled by multiple cell signaling pathways, including the mitogen-activated protein kinase (MAPK)/ERK pathway,38, 39, 40 and they have been found to be increased in neurodegenerative diseases including AD, PD and ALS.25, 28, 32, 41, 42, 43, 44, 45, 46 Importantly, COX-2 inhibitors such as celecoxib exhibited significant neuroprotective effects and prolonged survival or delayed disease onset in a SOD1-ALS transgenic mouse model through the downregulation of PGE2 release.28
Most recent studies have tried to elucidate the role of glial cells in neurotoxicity using TDP-43-ALS models, which are considered to be helpful for better understanding the disease mechanisms.47, 48, 49, 50, 51 Although the contribution of glial cells to TDP-43-mediated motor neuron degeneration is now well supported, this model does not fully suggest an astrocyte-based non-cell autonomous mechanism. For example, recent studies have shown that TDP-43-mutant astrocytes do not affect the survival of motor neurons,50, 51 indicating a previously unrecognized non-cell autonomous TDP-43 proteinopathy that associates with cell types other than astrocytes.
Given that the role of glial cell types other than astrocytes in TDP-43-mediated neuroinflammation is still not fully understood, we aim to compare the contribution of microglia and astrocytes to neurotoxicity in a TDP-43 loss-of-function model. Here, we show that TDP-43 has a dominant role in promoting COX-2-PGE2 production through the MAPK/ERK pathway in primary cultured microglia, but not in primary cultured astrocytes. Our study suggests that overproduction of PGE2 in microglia is a novel molecular mechanism underlying neurotoxicity in TDP-43-linked ALS. Moreover, our data identify celecoxib as a new potential effective treatment of TDP-43-linked ALS and possibly other types of ALS.
Results
Selective upregulation of COX-2 in TDP-43-deficient microglia but not astrocytes
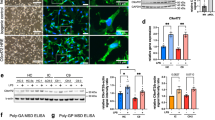
The goal of this study is to better understand the role of TDP-43 in neuroinflammation, a key feature of ALS. To gain insight into the roles of microglia and astrocytes in TDP-43-mediated neurotoxicity, we knocked down TDP-43 in primary cultured microglia and astrocytes using siRNA against TDP-43, and we then used biochemical approaches to detect the expression levels of COX-2 and iNOS, two markers of neuroinflammation. RNAi-mediated depletion of TDP-43 in microglia resulted in a greatly increased protein level of COX-2 with or without LPS treatment, whereas iNOS protein level was unchanged (Figure 1a). In contrast, protein levels of either COX-2 or iNOS were not increased in TDP-43-depleted astrocytes with or without LPS treatment (Figure 1b). To further explore the effects of regulating TDP-43 on the expression of COX-2 and iNOS, we performed quantitative real-time PCR (qRT-PCR) assays: the mRNA level of COX-2, but not iNOS, was remarkably upregulated in TDP-43-depleted microglia (Figure 1c). Similar results were obtained in BV2 cells (microglial cell line) as in primary cultured microglia (Supplementary Figure S1). Taken together, these results indicate a critical role for TDP-43 in COX-2 expression in microglia.

TDP-43 specifically regulates the expression level of COX-2, but not iNOS, in microglia. (a) Primary cultured microglia were transfected with si-control and si-TDP-43. After 72 h, the cells were treated with PBS or LPS (0.1 μg/ml) for 24 h, and then the cell lysates were subjected to immunoblot analysis using antibodies targeting iNOS, COX-2, TDP-43 and GAPDH. The quantification of TDP-43, COX-2 and iNOS levels is shown in the lower panels, representing three independent experiments. The data are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA. (b) Experiments similar to those in (a) were performed in primary cultured astrocytes. The quantification of TDP-43, COX-2 and iNOS levels is shown on the lower side. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA. (c) Primary cultured microglia were transfected with si-control and si-TDP-43 and cultured for 96 h, and then the cells were processed for qRT-PCR. The mRNA levels of COX-2 and iNOS were each quantified and normalized to GAPDH. The data from three independent experiments are shown as the means±S.E.M., ns, not significantly different; **P<0.01; one-way ANOVA
TDP-43 specifically regulates COX-2 expression in a MAPK/ERK-dependent manner
To characterize the molecular mechanism underlying the upregulation of COX-2 in TDP-43-deficient microglia, we examined the changes in cell signaling pathways accompanied by upregulation of COX-2 expression in TDP-43-deficient microglia. Given that the classical MAPK signaling pathway could regulate COX-2 expression and is significantly associated with various neurodegenerative diseases, we hypothesized that TDP-43 might be involved in the regulation of MAPKs. To test this possibility, we detected the total and phosphorylated levels of MEK, ERK, JNK and p38 in TDP-43-deficient microglia. As shown in Figures 2a and b, depletion of TDP-43 markedly increased phosphorylated MEK and ERK but not phosphorylated JNK or p38. Meanwhile, total protein levels of JNK, p38, MEK and ERK were not changed.

Loss of TDP-43 enhanced COX-2 expression by specifically activating MAPK/ERK signaling. (a) BV2 cells were transfected with si-control and si-TDP-43. At 72 h after transfection, the cell lysates were subjected to immunoblot analysis using anti-p-JNK1/2, JNK1/2, p-p38, p38, TDP-43 and GAPDH antibodies. The quantification data are shown on the lower side. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; one-way ANOVA. (b) Similar experiments as in (a) were performed, but using anti-p-MEK1/2, MEK1, p-ERK1/2, ERK1/2, TDP-43 and GAPDH antibodies. The relative densities are shown on the lower side. The data from three independent experiments are indicated as the means±S.E.M.; ns, not significantly different; *P<0.05; **P<0.01; one-way ANOVA. (c) BV2 cells were transfected with si-control and si-TDP-43 and cultured for 48 h. Then, the cells were treated with DMSO or U0126 (20 μM) for 24 h. Subsequently, the cells were processed for qRT-PCR analysis. The mRNA level of COX-2 was quantified and normalized to GAPDH. Data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA. (d) Similar transfections and treatments as in (c) were performed in BV2 cells, and the cell lysates were subjected to immunoblot analysis using anti-COX-2, p-ERK1/2, TDP-43 and GAPDH antibodies. The relative densities are shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA. (e–g) Similar experiments as in (d) were performed, but by incubating cells with SP600125 (10 μM), SB216763 (10 μM) and SB203580 (10 μM) instead of U0126. The cell lysates were subjected to immunoblot analysis using anti-COX-2, TDP-43, GAPDH, p-JNK1/2 and p-GS antibodies. The relative densities are shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; *P<0.05; **P<0.01; one-way ANOVA
Given that TDP-43 strongly enhanced MAPK/ERK activation and COX-2 expression, we hypothesized that TDP-43 might regulate COX-2 by targeting ERK. Relatedly, we found that both mRNA and protein levels of COX-2 were no longer increased in TDP-43-deficient microglia under treatment with U0126, a MEK1 inhibitor (Figures 2c and d). In contrast, treatments with a set of other inhibitors, SP600125 (JNK inhibitor), SB216763 (GSK3β inhibitor) and SB203580 (p38 inhibitor), had no effect on TDP-43-mediated COX-2 expression (Figures 2e and g). Therefore, our data demonstrate that TDP-43 regulates microglial COX-2 expression by specifically targeting MAPK/ERK and not other cell signaling pathways.
TDP-43 regulates AP-1 transcriptional activity by targeting the MAPK/ERK pathway
Among the various signaling molecules associated with COX-2 expression, NF-κB and AP-1 are two transcription factors that have been known to act downstream of MAPK/ERK, and they can tightly regulate COX-2 expression. To identify which signaling pathway is involved in TDP-43-mediated COX-2 expression, we first employed an NF-κB inhibitor, BAY. The protein expression level of COX-2 was still increased in TDP-43-deficient microglia treated with BAY (Figure 3a), suggesting that TDP-43 regulates COX-2 in an NF-κB-independent manner. Next, we detected the transcriptional activity of AP-1 in TDP-43-depleted cells using a luciferase reporter gene assay. We found that depletion of TDP-43 significantly increased AP-1 transcriptional activity in microglia under normal conditions but not under treatment of U0126 (Figures 3b and c), suggesting that TDP-43 regulates AP-1 transcriptional activity in an ERK-dependent manner.

TDP-43 influences AP-1 transcriptional activity in a MAPK-signaling-dependent manner. (a) Experiments similar to those in Figure 2d were performed in BV2 cells, but by incubating cells with BAY (25 μM) instead of U0126. The cell lysates were subjected to immunoblot analysis using anti-COX-2, TDP-43 and GAPDH antibodies. The relative densities are shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; *P<0.05; **P<0.01; one-way ANOVA. (b) The stable BV2 AP-1 cell lines were transfected with si-control and si-TDP-43 for 72 h. Then, the expression of the AP-1 reporter gene was assessed. Data from three independent experiments are presented as the means±S.E.M.; **P<0.01; one-way ANOVA. (c) Similar experiments as in (b) were performed, and the cells were treated with DMSO or U0126 (20 μM) for the last 24 h of the transfection. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA
TDP-43 controls the release of PGE2 in a MAPK/ERK- and COX-2-dependent manner
Because COX-2 activity is strongly associated with the production of PGE2, a key downstream product of COX-2, we next investigated whether TDP-43 could regulate the production of PGE2. We found that depletion of TDP-43 greatly increased the release of PGE2 from microglia, but not from astrocytes, in a time-dependent manner (Figures 4a and b, and Supplementary Figure S2). However, this effect of TDP-43 was blocked by incubating the microglia with either U0126 or celecoxib, a specific COX-2 inhibitor (Figures 4c and d). Taken together, our results suggest a key role for COX-2 in TDP-43-mediated regulation of PGE2.

Loss of TDP-43 strikingly promotes the production of PGE2 in microglia but not in astrocytes. (a) Primary cultured microglia were transfected with si-control and si-TDP-43 for 96 h. Then, the media from for 24 h cultures of microglia were subjected to PGE2 enzyme-linked immunosorbent assays (ELISAs). The data from three independent experiments are presented as the means±S.E.M.; **P<0.01; one-way ANOVA (b) BV2 cells and primary cultured astrocytes were transfected with si-control or si-TDP-43 for 72 h or 96 h at a final density of 1 × 106 cells per well in 12-well plates. Then, the 1 : 10 diluted media from 24 h cultures of BV2 cells or primary cultured astrocytes were subjected to PGE2 ELISAs. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA. (c) BV2 cells were transfected with si-control or si-TDP-43 for 48 h. Then, the cells were treated with DMSO or U0126 (20 μM) for 24 h, with a final cell density of 3 × 105 cells per well in 24-well plates. Next, the media from 24 h cultures of BV2 cells were subjected to PGE2 ELISAs. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA. (d) Similar experiments as in (b) were performed, but the BV2 cells were treated with celecoxib (50 μM) instead of U0126. The data from three independent experiments are indicated as the means±S.E.M.; ns, not significantly different; **P<0.01; one-way ANOVA
Loss of TDP-43 induces selective microglia-mediated neuronal death
Based on the previous observations that increased levels of PGE2 were toxic to neurons, we hypothesized that loss of TDP-43 in microglia could initiate neuronal death owing to the overproduction of PGE2. To test this possibility, we cultured neurons in media collected from TDP-43-depleted microglia (conditioned medium assay) (Figures 5a and c), and the conditioned medium significantly induced cell death in both cortical neurons (Figures 5b and Supplementary Figure S3A) and motor neurons (Figures 5d and Supplementary Figure S3B), but not in mouse embryonic fibroblasts (Figures 5g and h and Supplementary Figure S3C). Interestingly and importantly, the culture medium from TDP-43-depleted astrocytes had no effect on neuron viability (Figures 5e and f).

The culture medium from TDP-43-depleted microglia, but not TDP-43-depleted astrocytes, is toxic to neurons. (a) Scheme of the conditioned medium assay using TDP-43-depleted primary cultured microglia and cortical neurons. (b) Primary cultured microglia were transfected with si-control and si-TDP-43 for 96 h. The culture medium from the last 24 h was harvested and used to culture primary cortical neurons transfected with lentiviral EGFP for 16 h. Then, the cells were visualized using confocal microscopy (Zeiss LSM 710). Scale bars, 30 μm, upper panel; 10 μm, lower panel. The percentage of viability among cortical neurons is shown on the lower side. The data from three independent experiments are presented as the means±S.E.M.; **P<0.01; one-way ANOVA. (c) Scheme of primary cultured microglia culture medium treatment of primary cultured motor neurons. (c and d) Similar experiments as in (a and b) were performed in primary cultures of motor neurons instead of cortical neurons. The cells were fixed, stained with an antibody against MAP2 (neuronal marker) and visualized using fluorescence microscopy (Olympus IX71). Scale bar, 10 μm. The percentage of motor neurons that were viable is shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; **P<0.01; one-way ANOVA. (e and f) Similar experiments as in (a and b) were performed, using primary cultured astrocytes instead of primary cultured microglia. The cells were visualized using confocal microscopy. Scale bars, 30 μm, upper panel; 10 μm, lower panel. The percentage of cortical neurons that were viable is shown on the lower side. The data from three independent experiments indicated the means±S.E.M., ns, not significantly different, one-way ANOVA. (g and h) Similar experiments as in (a and b) were performed, using mouse embryonic fibroblasts instead of primary cortical neurons. The cells were visualized using confocal microscopy. Scale bars, 30 μm, upper panel; 10 μm, lower panel. The percentage of primary mouse embryonic fibroblasts that were viable is shown on the lower side. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; one-way ANOVA
Celecoxib alleviates neuronal death induced by TDP-43-depleted microglia
We next investigated whether the COX-2 inhibitor celecoxib could alleviate neuronal death driven by TDP-43-depleted microglia. We performed the conditioned medium assay as described in Figures 5a and c, and we found that the impaired viability of both cortical neurons and motor neurons were restored upon incubating TDP-43-depleted microglia with celecoxib (Figures 6a and c). Meanwhile, celecoxib alone had no effect on the viability of cortical or motor neurons (Figures 6d and e).

Celecoxib alleviates neuronal death mediated by TDP-43-deficient microglia. (a and b) BV2 cells were transfected with si-control and si-TDP-43. After 72 h, BV2 cells were treated with DMSO or celecoxib for another 24 h. The culture media from the last 24 h were harvested and used to culture primary cortical neurons transfected with lentiviral EGFP for 8 h (the conditioned medium assay as described for Figure 5). Then, the cells were visualized using confocal microscopy. Scale bars, 30 μm (a); 10 μm (b). The percentage of cortical neurons that were viable is shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; **P<0.01; one-way ANOVA. (c) Similar experiments as in (a and b) were performed in primary cultured motor neurons. The cells were fixed, stained with an antibody against MAP2 and visualized using fluorescence microscopy. Scale bar, 10 μm. The percentage of motor neurons that were viable is shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; **P<0.01; one-way ANOVA. (d) Primary cultured cortical neurons were transfected with lentiviral EGFP. After 72 h, the cortical neurons were treated with DMSO or celecoxib for another 24 h. Then, the cells were visualized using confocal microscopy. Scale bars, 30 μm, upper panel; 10 μm, lower panel. The percentage of cortical neurons that were viable is shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; one-way ANOVA. (e) Similar experiments as in (d) were performed in primary cultured motor neurons instead of primary cultured cortical neurons. The cells were fixed, stained with an antibody against MAP2, and visualized using fluorescence microscopy. Scale bar, 10 μm. The percentage of motor neurons that were viable is shown on the right side. The data from three independent experiments are presented as the means±S.E.M.; ns, not significantly different; one-way ANOVA
Discussion
Although initially, most studies on ALS focused on the selective loss of motor neurons themselves, increasing efforts to understand the role of glial cells in disease pathogenesis have come to the forefront of the field. The present study reveals microglia-mediated COX-2-PGE2 production as the molecular determinant of TDP-43-associated neurotoxicity, emphasizing the important contribution of microglia to non-cell-autonomous motor neuron degeneration in TDP-43-linked ALS (Figure 7).

Neuronal death is mediated by the classical MAPK signaling pathway and upregulated PGE2 in TDP-43-depleted microglia. The schematic illustrates the mechanism by which TDP-43-depleted microglia, but not TDP-43-depleted astrocytes, induce neuronal death. The MAPK/ERK signaling pathway is activated in TDP-43-depleted microglia, resulting in increased COX-2 and PGE2, which then trigger neuronal death. Inhibition of COX-2 and PGE2 production using celecoxib alleviates neuronal death mediated by TDP-43-depleted microglia
Although numerous reports have shown that astrogliosis is associated with disease progression, ALS astrocytes carrying pathogenic mutant TDP-43 did not exhibit toxicity to motor neurons in the short term.51 On the basis of this finding, it is possible that other types of cells surrounding motor neurons are involved in the initial toxic effect of motor neurons. Consistent with this notion, microglia have recently been suggested to have a role in ALS initiation,52 but the contribution of microglia to TDP-43-linked ALS is still not fully understood. In this study, we show that loss of TDP-43 in microglia strikingly triggers the increase of COX-2 (roughly threefold, Figure 1a), accelerating the microglial inflammatory reaction through the release of PGE2 (roughly 10-fold, Figure 4), and mediates selective neuronal death (Figure 5). Because the level of COX-2 in astrocytes is much lower than that in microglia (Figures 1a and b, shown in the immunoblots), we reason that in astrocytes, it would be difficult to increase the expression of COX-2 to levels comparable to those in microglia through the loss of TDP-43, thus, the production of PGE2 in astrocytes would not be sufficient to cause neuronal death (Figure 1b,4b and 5f). Despite that, an interesting study showed that astrogliosis could induce motor neuron death in a rat model with selective overexpression of mutant TDP-43 in astrocytes,47 we used a TDP-43 loss-of-function model instead of a TDP-43 gain-of-function model in the current study, and our data showed that loss of TDP-43 in astrocytes is not sufficient to enhance COX-2-PGE2 production, and is thus not sufficient to trigger neurotoxicity. It is therefore possible that the regulation of neuroinflammation may vary in different TDP-43-linked ALS models. Interestingly and importantly, a very recent study provides evidence that microglia specifically induced neurotoxicity via NF-κB activation in a mutant SOD1 transgenic mouse model of ALS. In addition, inhibition of NF-κB in microglia, but not astrocytes, rescued the survival of motor neurons in vitro and extended survival in SOD1 mice.53 Together with our study, this suggests that microglia may have a key, non-cell-autonomous role in ALS pathogenesis, and it provides novel insight into potential therapeutic treatment of ALS by targeting microglia. Although further investigations are needed to better understand the role of microglia in TDP-43-mediated ALS using better models such as animal models or iPSC-derived cells from ALS patients, a remarkable concordance between the gene expression profile of in vitro co-cultured astrocytes with motor neurons carrying mutant SOD1 and spinal cords of mutant SOD1 transgenic mice has been found in a recent study,54 suggesting that our study using cultured cell model is highly relevant to in vivo ALS research.
Because cytoplasmic TDP-43 aggregates accompanied by a loss of nuclear TDP-43 have been found in ALS patients, a major unresolved question regarding TDP-43-mediated neurodegeneration is that whether the toxicity is triggered by a toxic gain-of-function or by a loss-of-function. Consistent with a gain-of-function mechanism, several cellular signaling pathways, such as PTEN, insulin/IGF-1 and redox signaling, have been reported to regulate TDP-43 in models expressing mutant TDP-43;55, 56, 57, 58 consistent with a loss-of-function mechanism, TDP-43 participates in the regulation of the heme oxygenase-1, Rac1-AMPAR and JNK pathways.59, 60, 61 However, there is increasing evidence that loss-of-function, rather than gain-of-function, is the major mechanism mediating TDP-43 neuropathology.5, 12, 13, 14 Thus, here, we analyzed multiple cellular signaling pathways, including MAPK, JNK, p38 and GSK3β, in TDP-43-depleted cells, and we confirmed that MEK-ERK signaling was specifically upregulated in microglia with TDP-43 knockdown (Figures 2a and b). Given that COX-2 expression is controlled by NF-κB and AP-1, two transcription factors that function downstream of MEK-ERK signaling,62, 63 we thought to test whether NF-κB or AP-1 was involved in TDP-43-mediated regulation of COX-2. Our data indicate that NF-κB was not involved in this regulation because blocking NF-κB activity does not change the effect of TDP-43 on COX-2 expression (Figure 3a). Although a previous study showed that TDP-43 is associated with NF-κB activation and inflammation,64 it should be noted that TDP-43 itself did not regulate NF-κB activation and inflammation in their observations.64 It is possible that other inflammatory inducers and stimuli may help to trigger NF-κB-mediated inflammation in TDP-43-depleted microglia. In the current study, we find that TDP-43 can directly regulate COX-2-PGE2 production (without extra stimuli), indicating that signaling molecules other than NF-κB are required for this regulation. Relatedly, our results show that knockdown of TDP-43 in microglia resulted in the activation of AP-1 (Figure 3b) and led to marked increases in both PGE2 and COX-2. Moreover, inhibition of MEK-ERK signaling by U0126 strikingly diminished the abnormal increases in AP-1 activity, COX-2 expression and PGE2 production in TDP-43-deficient microglia (Figures 2d,3c and 4c). Taken together, these data reveal that the abnormal activation of MEK-ERK-AP-1 signaling is directly associated with the upregulation of COX-2-PGE2 production in TDP-43-depleted microglia.
We found increased COX-2 expression and PGE2 production in TDP-43-deficient microglia (Figures 1a and 4a), and inhibition of COX-2 expression and PGE2 production by celecoxib treatment reduced the neurotoxicity triggered by TDP-43-deficient microglia (Figures 6a and c). Consistent with our observations, previous studies have shown that inhibition of COX-2 and PGE2-mediated inflammation was therapeutically effective in mutant SOD1 transgenic mice.28, 65 Given that abnormal TDP-43 function is tightly associated with most ALS cases, the present study provides novel insight into how microglia induce neurotoxicity in ALS, and, more interestingly, suggests celecoxib as a potential therapy for ALS.
Materials and Methods
Antibodies
The anti-TDP-43 antibody was described previously,66 and the following additional primary antibodies were used: anti-iNOS (Abcam, Cambridge, MA, USA), anti-COX-2 (Epitomics, Burlingame, CA, USA), anti-GAPDH (Chemicon, Temecula, CA, USA), anti-phospho-MEK (S217/221) (Cell Signaling Technology, Beverly, MA, USA), anti-MEK (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho-ERK (Santa Cruz Biotechnology), anti-ERK (Santa Cruz Biotechnology), anti-phospho-JNK (Epitomics), anti-JNK (Santa Cruz Biotechnology), anti-phospho-p38 (Epitomics), anti-p38 (Santa Cruz Biotechnology), anti-phospho-c-Raf (S338) (Cell Signaling Technology), anti-c-JUN (Proteintech, Chicago, IL, USA), anti-phospho-Glycogen Synthase (S641) (p-GS) (Epitomics) and anti-MAP2 (Santa Cruz Biotechnology). The secondary antibodies used included horseradish peroxidase-conjugated sheep anti-mouse and anti-rabbit antibodies (Amersham Pharmacia Biotech, Peapack, NJ, USA). The antibody-bound proteins were visualized using an ECL detection kit (Amersham Biosciences, Piscataway, NJ, USA) or Alexa Fluor 488 (green) donkey anti-mouse IgG (Invitrogen, La Jolla, CA, USA).
Cell culture, transfection, drug treatment, and RNAi and cell viability
Mouse motor neuron cell line (NSC-34 cells) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) (Gibco) with penicillin (100 mg/ml) and streptomycin (100 mg/ml). Mouse microglial cell line (BV2 cells) were cultured in similar culture medium, but using 10% heat-inactivated FBS instead. For RNAi experiments, cells were transfected with siRNA against TDP-43 using the RNAiMAX (Invitrogen) transfection reagent according to the manufacturer’s instructions. Subsequently, cells were treated for 24 h with DMSO (Sangon Biotech, Shanghai, China) or U0126 (Beyotime, Shanghai, China) (20 μM), BAY (Beyotime) (25 μM), SP600125 (10 μM), SB216763 (Sigma, St. Louis, MO, USA) (10 μM), SB203580 (10 μM), celecoxib (Sigma) (50 μM), or PBS (Gibco) or LPS (Beyotime) (0.1 μg/ml). The following sequences were used for siRNA targeting mouse TDP-43 (si-TDP-43): 5′-GGATCTGAAAGACTATTTC-3′; siRNA targeting rat TDP-43 (si-TDP-43): 5′-CCAATGCTGAACCTAAGCA-3′. For cell viability analysis, the cells were stained with PI and visualized using fluorescence microscopy. The dead cells with PI-positive staining were counted and the percentage of viability of cells was quantified.
Primary culture of microglia, astrocytes, cortical neurons, spinal cord motor neurons and mouse embryonic fibroblasts
Primary cultured microglia were prepared as described elsewhere.67 In brief, the cortex of new born SD rats was chopped and dissected. Cortex tissues were incubated with 0.25% Trypsin (Gibco) for 15 min at 37 °C, and then the cells were dissociated with a plastic pipette. Subsequently, cells isolated from cortex tissue were plated on poly-D-lysine-coated 24-well plates (Corning, Tewksbury, MA, USA) and cultured at 37 °C, 5% CO2. Mixed glial cells were cultured in DMEM/F12 with 10% heat-inactivated FBS and penicillin (100 mg/ml) and streptomycin (100 mg/ml), and the culture medium were replaced every 3 days. After 14 days, microglia were separated from mixed glial cells by shaking at 150 rpm for 2 h at 37 °C. Primary cultured astrocytes were similarly dissociated from the cortex of new born ICR mice and cultured in DMEM with 10% FBS and penicillin (100 mg/ml) and streptomycin (100 mg/ml). Cortical neurons were similarly dissociated from the cortex of ICR mouse embryos at embryonic day 17 (E17), and motor neurons were similarly dissociated from the spinal cord of ICR mouse embryos at E17 as previously described.68 In brief, the dissociated neurons were cultured in neurobasal medium (Gibco) with 10% FBS, 1 × B27 (Gibco) and glutamine (0.05 mg/ml; Sigma) for 12 h. Subsequently, culture medium were change to neurobasal medium with 1 × B27 and glutamine (0.05 mg/ml). After 5 days, the neurons were subjected to experiments. The purity of motor neurons were shown in Supplementary Figure S4A using specific markers of neurons (MAP2) and astrocytes (GFAP). Mouse embryonic fibroblasts were obtained from the epithelium of ICR mouse embryos at E17, and cultured as described for primary cultured astrocytes.
Immunoblot
Cells were harvested and lysed in cell lysis buffer (50 mM Tris-HCl (pH 7.6) with protease inhibitor cocktail (Roche, Indianapolis, IN, USA), 150 mM NaCl, 0.5% sodium deoxycholate, and 1% Nonidet P-40). Then, the proteins were separated by 10% or 12% SDS-PAGE (polyacrylamide gel electrophoresis) and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA).
QRT-PCR
Total RNA from BV2 cells was extracted with TRIzol Reagent (Invitrogen); subsequently, the RNA was reverse-transcribed into cDNA using PrimeScript RT Master Mix (Takara, Shiga, Japan). Real-time PCR analysis was performed using SYBR Green Real-Time PCR Master Mix (Takara) using a CFX96 Real-Time System (BIO-RAD, Hercules, CA, USA) and the following primers: mouse β-actin: 5′-GACCTGACTGACTACCTC-3′ and 5′-GACAGCGAGGCCAGGATG-3′, mouse iNOS: 5′-TCCCAGCCTGCCCCTTCAAT-3′ and 5′-CGGATCTCTCTCCTCCTGGG-3′, and mouse COX-2: 5′-CAGGCTGAACTTCGAAAC A-3′ and 5′-GCTCACGAGGCCACTGATACCTA-3′.
Analysis of PGE2 production
Primary cultured microglia and BV2 cells were transfected with siRNA against TDP-43. After transfection, the cells were treated with U0126 or celecoxib for 24 h, and then the levels of PGE2 in 50 μl of 400 μl medium were measured with PGE2 ELISA kits (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer’s instructions.
Luciferase reporter gene assay
BV2 cells were plated on 24-well plate and incubated for 16 h at 37 °C. Then the cells were transfected with Cignal lentiviral AP-1 Reporter (luc) (QIAGEN, Hilden, Japan) according to the manufacturer’s instructions for 24 h. Subsequently, the culture medium were replaced with fresh medium. After another 24 h, the cells were cultured in culture medium supplement with 2.5 μg/ml of puromycin. After 5 days, survived BV2 cells were selected and the BV2 stable cell line expressing AP-1 reporter construct was generated. Subsequently, the BV2 cells stably expressing AP-1 constructs were transfected with siRNA against TDP-43. After 48 h, the cells were treated with U0126 for 24 h, and then the cells were harvested. Finally, the AP-1 promoter activity was measured using the luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Abbreviations
- ALS:
-
amyotrophic lateral sclerosis
- AD:
-
Alzheimer’s disease
- COX-2:
-
cyclooxygenase-2
- DMSO:
-
dimethyl sulfoxide
- EGFP:
-
enhanced green fluorescent protein
- FBS:
-
fetal bovine serum
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- FTLD:
-
frontotemporal lobar degeneration
- MAPK:
-
mitogen-activated protein kinase
- PGE2:
-
prostaglandin E2
- PD:
-
Parkinson’s disease
- qRT-PCR:
-
quantitative real-time PCR
- siRNA:
-
small interfering RNA
- SOD1 :
-
Cu/Zn superoxide dismutase 1
- TARDBP :
-
TAR DNA-binding protein 43
References
Ling SC, Polymenidou M, Cleveland DW . Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013; 79: 416–438.
Lagier-Tourenne C, Cleveland DW . Rethinking ALS: the FUS about TDP-43. Cell 2009; 136: 1001–1004.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314: 130–133.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319: 1668–1672.
Vanden Broeck L, Callaerts P, Dermaut B . TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends Mol Med 2014; 20: 66–71.
Lagier-Tourenne C, Polymenidou M, Cleveland DW . TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 2010; 19 (R1): R46–R64.
Mackenzie IR, Rademakers R, Neumann M . TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010; 9: 995–1007.
Lippa CF, Rosso AL, Stutzbach LD, Neumann M, Lee VM, Trojanowski JQ . Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol 2009; 66: 1483–1488.
Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol 2014; 127: 811–824.
Chanson JB, Echaniz-Laguna A, Vogel T, Mohr M, Benoilid A, Kaltenbach G et al. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson's disease. Neurodegener Dis 2010; 7: 260–264.
Markopoulou K, Dickson DW, McComb RD, Wszolek ZK, Katechalidou L, Avery L et al. Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson's disease. Variability in familial Parkinson's disease. Acta Neuropathol 2008; 116: 25–35.
Xu ZS . Does a loss of TDP-43 function cause neurodegeneration? Mol Neurodegener 2012; 7: 27.
Vanden Broeck L, Naval-Sanchez M, Adachi Y, Diaper D, Dourlen P, Chapuis J et al. TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Rep 2013; 3: 160–172.
Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJ, Stepto A et al. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet 2013; 22: 1539–1557.
Ilieva H, Polymenidou M, Cleveland DW . Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 2009; 187: 761–772.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH . Mechanisms underlying inflammation in neurodegeneration. Cell 2010; 140: 918–934.
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006; 312: 1389–1392.
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K . Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 2007; 10: 608–614.
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC . Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008; 3: 637–648.
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 2007; 10: 615–622.
Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 2011; 29: 824–828.
Wang L, Gutmann DH, Roos RP . Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet 2011; 20: 286–293.
Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP et al. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol 2001; 49: 176–185.
Yasojima K, Tourtellotte WW, McGeer EG, McGeer PL . Marked increase in cyclooxygenase-2 in ALS spinal cord: implications for therapy. Neurology 2001; 57: 952–956.
Almer G, Teismann P, Stevic Z, Halaschek-Wiener J, Deecke L, Kostic V et al. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology 2002; 58: 1277–1279.
Ilzecka J . Prostaglandin E2 is increased in amyotrophic lateral sclerosis patients. Acta Neurol Scand 2003; 108: 125–129.
Maihofner C, Probst-Cousin S, Bergmann M, Neuhuber W, Neundorfer B, Heuss D . Expression and localization of cyclooxygenase-1 and -2 in human sporadic amyotrophic lateral sclerosis. Eur J Neurosci 2003; 18: 1527–1534.
Klivenyi P, Kiaei M, Gardian G, Calingasan NY, Beal MF . Additive neuroprotective effects of creatine and cyclooxygenase 2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem 2004; 88: 576–582.
Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol 2006; 6: 12.
Feng ZH, Wang TG, Li DD, Fung P, Wilson BC, Liu B et al. Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1, 2, 3, 6-tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci Lett 2002; 329: 354–358.
Pasinetti GM, Aisen PS . Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience 1998; 87: 319–324.
Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S et al. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci USA 2003; 100: 5473–5478.
Miyagishi H, Kosuge Y, Yoneoka Y, Ozone M, Endo M, Osada N et al. Prostaglandin E2-induced cell death is mediated by activation of EP2 receptors in motor neuron-like NSC-34 cells. J Pharmacol Sci 2013; 121: 347–350.
Thirumangalakudi L, Rao HV, Grammas P . Involvement of PGE2 and PGDH but not COX-2 in thrombin-induced cortical neuron apoptosis. Neurosci Lett 2009; 452: 172–175.
Takadera T, Shiraishi Y, Ohyashiki T . Prostaglandin E2 induced caspase-dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int 2004; 45: 713–719.
Jonakait GM, Ni L . Prostaglandins compromise basal forebrain cholinergic neuron differentiation and survival: action at EP1/3 receptors results in AIF-induced death. Brain Res 2009; 1285: 30–41.
Carrasco E, Casper D, Werner P . PGE(2) receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE(2) neurotoxicity. J Neurosci Res 2007; 85: 3109–3117.
McElroy SJ, Hobbs S, Kallen M, Tejera N, Rosen MJ, Grishin A et al. Transactivation of EGFR by LPS induces COX-2 expression in enterocytes. PLoS One 2012; 7: e38373.
Benoit V, Relic B, Leval Xd X, Chariot A, Merville MP, Bours V . Regulation of HER-2 oncogene expression by cyclooxygenase-2 and prostaglandin E2. Oncogene 2004; 23: 1631–1635.
Sudbo J . Novel management of oral cancer: a paradigm of predictive oncology. Clin Med Res 2004; 2: 233–242.
Knott C, Stern G, Wilkin GP . Inflammatory regulators in Parkinson's disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci 2000; 16: 724–739.
Mattammal MB, Strong R, Lakshmi VM, Chung HD, Stephenson AH . Prostaglandin H synthetase-mediated metabolism of dopamine: implication for Parkinson's disease. J Neurochem 1995; 64: 1645–1654.
Abdel-Halim MS, Lunden I, Cseh G, Anggard E . Prostaglandin profiles in nervous tissue and blood vessels of the brain of various animals. Prostaglandins 1980; 19: 249–258.
Seregi A, Forstermann U, Heldt R, Hertting G . The formation and regional distribution of prostaglandins D2 and F2 alpha in the brain of spontaneously convulsing gerbils. Brain Res 1985; 337: 171–174.
Bazan NG, Colangelo V, Lukiw WJ . Prostaglandins and other lipid mediators in Alzheimer's disease. Prostaglandins Other Lipid Mediat 2002; 68-69: 197–210.
Ahmad AS, Yun YT, Ahmad M, Maruyama T, Dore S . Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotox Res 2008; 14: 343–351.
Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X et al. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 2013; 32: 1917–1926.
Diaper DC, Adachi Y, Lazarou L, Greenstein M, Simoes FA, Di Domenico A et al. Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum Mol Genet 2013; 22: 3883–3893.
Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci USA 2013; 110: 4069–4074.
Haidet-Phillips AM, Gross SK, Williams T, Tuteja A, Sherman A, Ko M et al. Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp Neurol 2013; 250: 250–259.
Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci USA 2013; 110: 4697–4702.
Gerber YN, Sabourin JC, Rabano M, Vivanco M, Perrin FE . Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PLoS One 2012; 7: e36000.
Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014; 81: 1009–1023.
Phatnani HP, Guarnieri P, Friedman BA, Carrasco MA, Muratet M, O'Keeffe S et al. Intricate interplay between astrocytes and motor neurons in ALS. Proc Natl Acad Sci USA 2013; 110: E756–E765.
Ayala V, Granado-Serrano AB, Cacabelos D, Naudi A, Ilieva EV, Boada J et al. Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: implications in ALS. Acta Neuropathol 2011; 122: 259–270.
Zhang T, Mullane PC, Periz G, Wang J . TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet 2011; 20: 1952–1965.
Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VM . Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J 2012; 31: 1241–1252.
Zheng M, Liao M, Cui T, Tian H, Fan DS, Wan Q . Regulation of nuclear TDP-43 by NR2A-containing NMDA receptors and PTEN. J Cell Sci 2012; 125 (Pt 6): 1556–1567.
Duan W, Li X, Shi J, Guo Y, Li Z, Li C . Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010; 169: 1621–1629.
Majumder P, Chen YT, Bose JK, Wu CC, Cheng WC, Cheng SJ et al. TDP-43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathol 2012; 124: 231–245.
Suzuki H, Matsuoka M . The JNK/c-Jun signaling axis contributes to the TDP-43-induced cell death. Mol Cell Biochem 2013; 372: 241–248.
Hu Q, Li B, Xu R, Chen D, Mu C, Fei E et al. The protease Omi cleaves the mitogen-activated protein kinase kinase MEK1 to inhibit microglial activation. Science Signal 2012; 5: ra61.
Hayashi G, Shen Y, Pedersen TL, Newman JW, Pook M, Cortopassi G . Frataxin deficiency increases cyclooxygenase 2 and prostaglandins in cell and animal models of Friedreich's ataxia. Hum Mol Genet 2014; 23: 6838–6847.
Swarup V, Phaneuf D, Dupre N, Petri S, Strong M, Kriz J et al. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med 2011; 208: 2429–2447.
Shin JH, Lee YA, Lee JK, Lee YB, Cho W, Im DS et al. Concurrent blockade of free radical and microsomal prostaglandin E synthase-1-mediated PGE2 production improves safety and efficacy in a mouse model of amyotrophic lateral sclerosis. J Neurochem 2012; 122: 952–961.
Wang X, Fan H, Ying Z, Li B, Wang H, Wang G . Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett 2010; 469: 112–116.
Tao L, Zhang F, Hao L, Wu J, Jia J, Liu JY et al. 1-O-tigloyl-1-O-deacetyl-nimbolinin B inhibits LPS-stimulated inflammatory responses by suppressing NF-kappaB and JNK activation in microglia cells. J Pharmacol Sci 2014; 125: 364–374.
Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E et al. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet 2009; 18: 4268–4281.
Acknowledgements
This work was supported in part by the National High-tech Research and Development program of China 973-projects (2011CB504102), the National Natural Sciences Foundation of China (Nos. 31330030, 31200803 and 31371072), Natural Science Foundation of Jiangsu Province (BK2012181), a Project Funded by Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psycho-Diseases (BM2013003) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International Licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xia, Q., Hu, Q., Wang, H. et al. Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis 6, e1702 (2015). https://doi.org/10.1038/cddis.2015.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.69
This article is cited by
-
Update on the pathological roles of prostaglandin E2 in neurodegeneration in amyotrophic lateral sclerosis
Translational Neurodegeneration (2023)
-
Inhibition of the MEK/ERK pathway suppresses immune overactivation and mitigates TDP-43 toxicity in a Drosophila model of ALS
Immunity & Ageing (2023)
-
Loss of DJ-1 function contributes to Parkinson’s disease pathogenesis in mice via RACK1-mediated PKC activation and MAO-B upregulation
Acta Pharmacologica Sinica (2023)
-
The neuroprotective effects of peracetylated chitosan oligosaccharides against β-amyloid-induced cognitive deficits in rats
Marine Life Science & Technology (2023)
-
The murine metastatic microenvironment of experimental brain metastases of breast cancer differs by host age in vivo: a proteomic study
Clinical & Experimental Metastasis (2023)
