
Abstract
Apoptosis has been established as a mechanism of anti-cancer defense. Members of the BCL-2 family are critical mediators of apoptotic cell death in health and disease, often found to be deregulated in cancer and believed to lead to the survival of malignant clones. However, over the years, a number of studies pointed out that a model in which cell death resistance unambiguously acts as a barrier against malignant disease might be too simple. This is based on paradoxical observations made in tumor patients as well as mouse models indicating that apoptosis can indeed drive tumor formation, at least under certain circumstances. One possible explanation for this phenomenon is that apoptosis can promote proliferation critically needed to compensate for cell loss, for example, upon therapy, and to restore tissue homeostasis. However, this, at the same time, can promote tumor development by allowing expansion of selected clones. Usually, tissue resident stem/progenitor cells are a major source for repopulation, some of them potentially carrying (age-, injury- or therapy-induced) genetic aberrations deleterious for the host. Thereby, apoptosis might drive genomic instability by facilitating the emergence of pathologic clones during phases of proliferation and subsequent replication stress-associated DNA damage. Tumorigenesis initiated by repeated cell attrition and repopulation, as confirmed in different genetic models, has parallels in human cancers, exemplified in therapy-induced secondary malignancies and myelodysplastic syndromes in patients with congenital bone marrow failure syndromes. Here, we aim to review evidence in support of the oncogenic role of stress-induced apoptosis.
Similar content being viewed by others


Facts
-
During cancer development, clonal selection is facilitated by the acquisition of mutations in oncogenes and tumor suppressors and by the selection of 'winner' cells.
-
Apoptosis of (pre)-cancerous cells generates vacant niches that potentially become repopulated by more aggressive sub-clones. Thereby, apoptosis increases proliferative pressure and promotes clonal selection, thus driving tumor evolution.
-
Dying cells can promote cell division of neighboring cells.
Open Questions
-
Does apoptosis drive malignant transformation in pre-malignant conditions such as therapy-related myelodysplastic syndromes or congenital bone marrow failure syndromes?
-
Can resistance to apoptosis delay the risk of (further) malignant transformation within fully established tumors or in pre-malignant tissues?
-
How can unnecessary tissue damage and inflammatory response be avoided in tumor patients as well as in patients presenting with premalignant conditions?
Two concepts in the field of tumorigenesis are widely accepted. First, cancer is the result of sequential genetic changes that, eventually, transform normal into malignant cells, a model that has been referred to as multistep carcinogenesis.1 Second, specific biological processes have to be deregulated during tumor evolution to enable and sustain tumorigenesis. These processes have been summarized as 'hallmarks of cancer' by Hanahan and Weinberg2 in the years 2000 and 2011, respectively, and among those, evasion from cell death is still regarded as an essential mechanism required for malignant transformation and tumor maintenance.
Though every dogma has its day, doubt is the driving force behind scientific progress. In this review, we challenge the current paradigmatic view that increased survival is unambiguously promoting tumorigenesis. We will discuss the role of apoptosis and its deregulation during the induction, progression and maintenance of malignant disease. Finally, we propose to adopt the current view that resistance to cell death constitutes a genuine hallmark of cancer, as we believe that this may actually be limited to certain settings.
Apoptosis deregulation in cancer
Though cells can commit suicide by multiple ways, most cell death in vertebrates is mediated by the mitochondrial (intrinsic) pathway that is initiated by a plethora of signals, such as DNA damage, growth factor deprivation, developmental cues as well as many standard anti-cancer therapies. The initiators of this pathway belong to the BCL-2 family (Figure 1) and the balance between anti- and pro-apoptotic family members, the so-called 'Bcl-2 rheostat' determines whether a cell will live or die an apoptotic death.

Cytotoxic agents impact the Bcl-2 rheostat. The pro- and anti-apoptotic BCL-2 family proteins closely interact at the mitochondrial membrane and regulate the intrinsic apoptosis pathway. Cellular stress causes activation of pro-apoptotic BCL-2 proteins from the BH3-only sub-group (BIM, PUMA etc.). These bind to and inhibit their anti-apoptotic antagonists (BCL-2, MCL-1 etc.), thus releasing and activating the downstream effectors BAK and BAX. Mitochondrial membrane permeabilization (MOMP) is triggered, and pro-apoptotic molecules released into the cytoplasm activate caspases (in more detail reviewed by Labi et al.120). Conventional cytotoxic agents interfere with upstream signaling pathways converging at the BCL-2 family level. In contrast, BH3-mimetics directly inhibit pro-survival BCL-2 proteins
On the basis of the 'hallmarks of cancer' concept mentioned above, Green and Evan3 proposed that the core changes converting a normal cell into a malignant one might be simply increased proliferation coupled to decreased cell death. Indeed, it is fully established that tumor cells dampen the apoptotic response, as only defective death prevents their effective elimination by intrinsic anti-cancer mechanisms or anti-cancer therapy.4
Progressive counteraction of pro-death signals is a consequence of exogenous as well as (epi)genetic changes in the rich set of factors regulating apoptosis. Though mutations in genes encoding for core components of the cell-death machinery, for example, BCL-2 family proteins, are described in literature, they are not particularly common. One prominent exception occurs in B-cell follicular lymphoma where fusions between BCL-2 and the immunoglobulin heavy chain gene are regularly detected thus raising BCL-2 protein levels.5, 6, 7 More commonly, upstream signaling pathways converging at the level of the BCL-2 family are deregulated within tumor cells secondarily leading to an imbalance in the BCL-2 rheostat tilting the balance in favor of survival.
The tumor suppressor gene TP53 is frequently mutated in human cancer and has early been linked to apoptosis.8, 9, 10 Activated upon DNA damage, hypoxia or oncogene activation, TP53 initiates cell cycle arrest and DNA repair or, in irreversibly damaged cells, senescence or apoptosis, with PUMA and NOXA being the main pro-apoptotic target genes.11, 12, 13, 14, 15 Loss-of-function mutations in TP53 result in apoptosis resistance and are frequently associated with advanced tumor stage and poor prognosis.16, 17 Besides TP53, other components of the DNA damage checkpoint pathway are frequently inactivated in human cancer (e.g., MDM2, ARF, RB1).18, 19 Common mutations in oncogenes causing their growth factor-independent activation can be found in the RAS-signaling pathway, such as mutations in the RAS genes itself (i.e., HRAS, NRAS, KRAS) or upstream/downstream components of growth factor signaling pathways (e.g., FLT3, CKIT, EGFR, PTPN11, CBL, NF1 etc.).20 The main BCL-2 players repressed downstream of these pathways are PUMA, BAD and BIM, BH3-only proteins that are kept inactive by AKT signaling.21, 22 In colorectal cancers, the adaptor protein paxillin has been reported to promote survival and chemo-resistance of tumor cells by increasing BCL-2 protein stability and abundance.23 The c-MYC-oncogene can induce tumors in its wild-type form and is overexpressed in most human tumor entities.24 Paradoxically, at high levels it promotes apoptosis involving BCL-2 family proteins. Many other mutated oncogenes or tumor suppressors have been described to deregulate intrinsic apoptosis signaling but detailed information on their main downstream apoptotic effectors is often lacking.
Besides cell-intrinsic pro-survival signals, support from the environment is critically required for tumor cell survival. For example, chronic lymphocytic leukemia cells survive well in a lymph-node niche in vivo but rapidly undergo apoptosis ex vivo. This indicates that (i) the downstream apoptosis machinery is usually functional in these cells, and, that (ii) they are ready-to-die and as such strictly dependent on niche-derived pro-survival signals.25 Indeed, high levels of the BH3-only proteins BIM, BMF and NOXA were detected in primary human chronic lymphocytic leukemia cells.26, 27, 28, 29 Co-culture with feeder or T-helper cells as well as addition of cytokines strongly delays ex vivo apoptosis by increasing expression of anti-apoptotic BCL-2 proteins.25, 30, 31
BCL-2 proteins as targets for anti-cancer therapies
Although resistance to cell death is an important feature of cancers, it is certainly not true that tumors are fully resistant to cell death.32 If true, anti-cancer therapies would invariably fail. The majority of today’s therapeutic regimens aim to directly kill tumor cells and often successfully reduces tumor mass. Depending on the treatment scheme, different but often overlapping pathways are engaged leading to apoptosis and/or necrosis (i.e., DNA damage, oxidative or metabolic stress, and others). Not unexpectedly, clinical trials use the amount of treatment-induced cell death within tumors to predict prognosis and decide on further treatment intensity (i.e., steroid-response in pediatric acute lymphoblastic leukemia).33
Most conventional cytotoxic agents act by activating the intrinsic apoptosis pathway (Figure 1). DNA damaging agents (i.e., etoposide or alkylating agents) as well as γ-irradiation induce apoptosis by TP53-mediated activation of PUMA and possibly also NOXA.12, 13 In contrast, steroids kill acute lymphoblastic leukemia cells by activation of BIM, PUMA and/or BMF,34 and imatinib has been shown to kill BCR-ABL-positive chronic lymphocytic leukemia cells in a BIM- and BAD-dependent manner.35 Alternatively, apoptosis susceptibility of chronic lymphocytic leukemia cells can be increased by the CXCR4 antagonist plerixafor because of mobilization-dependent loss of survival signals in lymph-node niches.25, 36 Thereby, pro-survival signals within tumor cells drop and the BCL-2 rheostat favors induction of apoptosis.
Recently, specific compounds that induce apoptosis directly at the BCL-2 level have been developed (Figure 1). These 'BH3-mimetics' (i.e., ABT-737, ABT-263, ABT-199 and Obatoclax) mimic BH3-activity by binding and inhibiting pro-survival BCL-2 proteins.37 They hold big promise for anti-cancer therapy, either alone or in combination with other modalities. Among all pro-survival BCL-2 proteins, only BCL-2, BCL-xL and BCL-W are bound with relevant affinities by ABT-737 and ABT-263.38, 39 Accordingly, resistance of tumor cells is commonly caused by high levels of MCL-1 and/or BFL1/A140 and thus targeting these two proteins has become increasingly interesting.
The apoptosis paradox in tumor development
Complete apoptosis resistance coupled with unleashed proliferation would make any tumor grow to a mass of unbearable size in a very short period of time, a fact inconsistent with the usually long latency of malignant disease. An uncontrollably proliferating cell has to undergo only 40 population doublings until a clinically detectable tumor mass comprising approximately 109 cells appears. This lesion would require only 10 further doublings to produce 1012 cells, the maximal tumor size compatible with human life.41 As such rapid growth is rare, evolving tumors must be characterized by a dynamic interplay between proliferation, cell death and/or senescence. This feature contributes to intra-clonal heterogeneity of tumors that consist of subpopulations of cells displaying variable rates of death, division and aggressiveness.42, 43
As discussed earlier, fully transformed cells might have acquired mutations increasing cell death thresholds and inhibiting their clearance.44 But how do apoptosis and acquired apoptosis-resistance actually impact on the process of malignant transformation? Is apoptosis resistance sufficient to transform a cell? When and why do transforming or transformed cells require mechanisms to evade apoptosis? And, are there situations where apoptosis resistance rather reduces the risk of (further) malignant transformation?
The involvement of BCL-2 itself in neoplastic transformation was nailed in 1985 when a translocation juxtaposing the BCL-2 gene and the immunoglobulin heavy chain gene t(14;18) was regularly found in human follicular lymphoma.5, 6, 7 This was the first evidence that some oncogenes rather promote cell survival than stimulate proliferation. Only later we learned that apoptosis inhibition per se is hardly ever sufficient to transform a cell, in line with the above-mentioned multi-step carcinogenesis model. BCL-2 transgenic mice develop tumors only at low penetrance and with long latency,45 and only a fraction of all persons harboring the t(14;18) translocation in blood cells subsequently develop follicular lymphoma, and only after a long-lasting latency period.46
The tumorigenic potential of BCL-2 becomes only evident when overexpressed in combination with oncogenes such as c-MYC. Though promoting unleashed proliferation, c-MYC can only efficiently immortalize cells in the presence of sufficient pro-survival signals such as those provided by overexpression of BCL-2.47, 48 This need arises because high levels of c-MYC drive cell death. Thus, apoptosis is an important barrier to uncontrolled proliferation and a form of tumor surveillance curtailing MYC-driven transformation. Consequent studies demonstrated that c-MYC synergizes with any of the anti-apoptotic BCL-2 proteins in transforming leukocytes in overexpression models in vivo,49 whereas the dependence appears more selective at the level of endogenous pro-survival proteins.50 Consistently, the loss of various pro-apoptotic BH3-only proteins results in acceleration of c-MYC-driven lymphomagenesis.51, 52, 53 Similar synergies between c-MYC and anti-apoptotic BCL-2 proteins have been observed in other tissues such as the pancreas or the mammary gland.54, 55 Furthermore, apoptosis is not only induced by c-MYC overexpression but also by activity-gain of other oncogenes or the loss of tumor suppressors. However, this is beyond the scope of this review and was discussed elsewhere.4, 56
Surprisingly, ample data from human tumors and mouse models actually indicate that the 'simple' view on apoptosis being a key mechanism of anti-cancer defense suffers from oversimplification. Studies on human tumors paradoxically pointed out a strong correlation between high BCL-2 levels and favorable prognosis (Table 1). These observations indicate that BCL-2 overexpression is routinely observed in human tumors and that it can be associated with a less aggressive disease course. Along that line, the increased expression of pro-apoptotic BAX has been correlated with an increased risk of relapse in childhood acute lymphoblastic leukemia.57 Gurova et al.58 demonstrated that clonal expansion of transformed TP53-deficient fibroblasts in vitro and in a mouse tumor model was suppressed by BCL-2 overexpression. Intriguingly, BCL-2-overexpressing tumors contained genetically stable cells and were able to restrict the expansion of otherwise rapidly growing and genetically instable TP53-deficient cells. In another study, BAX overexpression in the T-cell lineage enhanced lymphomagenesis in TP53-deficient mice in a dose-dependent manner, and even initiated lymphoma formation on a TP53-proficient background.59 In accordance with data discussed before, BAX-driven apoptosis led to increased chromosome instability, and co-expression of BCL-2 was able to delay lymphomagenesis.
In sum, these results support a hypothesis in which a higher rate of apoptosis within a tumor, either at early stage or during progression, or both, might promote genetic instability causing more aggressive disease.
A farewell from the classic view on the role of apoptosis in cancer
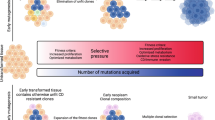
How could apoptosis promote tumorigenesis? Cancer development can be viewed as a Darwinistic process of somatic cell evolution, whereby initially 'healthy' cells acquire multiple (epi)genetic lesions driving clonal selection. This process is facilitated by the acquisition of mutations in oncogenes and tumor suppressors and by the selection for cells with superior fitness. Under continuous selection pressure, apoptosis could be a major driver of clonal expansion by generating vacant niches (Figure 2). These niches become repopulated by more aggressive sub-clones with certain competitive advantages. In that respect, apoptosis would be a driver of tumor evolution and a hallmark of aggressive disease. This could be especially relevant during early steps of tumorigenesis. Pre-malignant lesions can stably persist for an extended period of time while still too small to be clinically relevant.

How apoptosis shapes cancer. (a) In proliferative tissues, injury is followed by rapid regeneration and restoration of normally sized and shaped structures. In the Drosophila wing imaginal disc, apoptotic cells induce competitive proliferation by secretion of mitogenic factors in a caspase-dependent manner. (b) In tissues with aberrant cells, tissue injury (e.g. caused by DNA damage in MDS patients) and consecutive proliferation enables outgrowth of more aggressive clones. This fosters malignant transformation. (c) Within established tumors, chemo- or radiotherapy induces apoptosis but leads to death-induced proliferation of therapy-surviving cells. Following the generation of space, proliferation is mediated by mitogens derived from apoptotic cells (such as PGE2). As proposed in mathematical models, this results in increased sub-clonal variability with a higher risk of tumor progression, chemoresistance and relapse
Proof-of-principle experiments exploring cell competition and compensatory proliferation in D. melanogaster larval development suggest a dual role for apoptosis during early tumorigenesis with the need to dampen intrinsic pro-apoptotic signals to promote tumor cell survival on one hand, and the benefit from the death of surrounding cells on the other hand.60, 61 This can be described as a type of Darwinian-like selection that generates 'winner' and 'loser' cells thus leading to long-term outgrowth of certain cells over others. In particular, preventing apoptosis of surrounding wild-type cells impairs the growth of otherwise highly proliferating clones, both in the cases of DMYC-induced super-competition60 and Minute-induced cell-competition.61 It has only recently been shown that this competition relies on TOLL signaling inducing NFκB-dependent apoptosis in 'loser' cells and their subsequent engulfment by 'winner' cells.62, 63
Along this line, a natural cell competition has been described for thymic progenitor cells in the mouse. Young cells recently immigrated from bone marrow displace 'older' progenitors already residing in the thymus. The 'older', 'loser' cells, express lower BCL-2 levels and are more susceptible to apoptosis. Consequently, reduction of natural competition in healthy thymic tissue causes T-cell lymphoma.64, 65
Mathematical models allow for an approximation of how intrinsic cell properties influence growth dynamics and clonal expansion.66, 67 Enderling et al.68 predict that spontaneous cell death yields a tumor size reduction in the short term, but ultimately enhances tumorigenesis in the long term. They conclude that tumors can remain dormant for long intervals despite constant cellular turnover and that high apoptosis rates perturb the intrinsic tumor dynamics and shift the population towards more aggressive subclones.69 Wodarz et al.70 describe the relation between death rate and the generation of mutant cells within a population after a first wave of clonal expansion. In their mathematical model, they find that less cell death correlates with fewer cell divisions during clonal expansion, thus leading to a less variable cell population. In contrast, high death rates correlate with more cell divisions during expansion causing the appearance of many different mutants (Figure 2). With increasing sub-clonal variability, the risk that individual cells overcome selective barriers (i.e. growth inhibition) and progress towards malignancy increases.70 We are putting forward the question whether these in silico models on cell death-stimulated tumor progression find their counterparts in vivo.
Death-driven proliferation facilitates tissue regeneration and tumorigenesis
One piece of evidence in favor of these mathematical models comes from two studies on thymic T-cell lymphoma where genetic ablation of the pro-apoptotic BH3-only protein PUMA abolished tumor formation.71, 72 In this mouse model, lymphomagenesis is induced by repeated rounds of sub-lethal γ-irradiation and strongly accelerated by TP53 deficiency.73 Repeated γ-irradiation induces a massive wave of apoptosis in the hematopoietic compartment that is dependent on the TP53 target, PUMA.13 Initially, these studies aimed at confirming the tumor suppressor potential of PUMA, as was suggested by the fact that its loss accelerated MYC-induced lymphomagenesis.53, 74 Unexpectedly, PUMA deficiency protected mice efficiently from thymic lymphoma. Why does deletion of PUMA abrogate lymphoma formation whereas loss of its activator, TP53, does the opposite? Two studies on the competition of hematopoietic progenitors shed light on possibly underlying mechanisms. Hematopoietic stem cells (HSCs) carrying damaged DNA introduced by sub-lethal irradiation can effectively reconstitute myelo-ablated mice. However, they are outcompeted when transplanted in a competitive setting with TP53-deficient HSCs.75, 76 As transplantation of lethally irradiated recipients requires HSC expansion for hematopoietic regeneration, it is tempting to speculate that selective pressure during repopulation provides the basis for oncogenic mutations to appear. This theory is backed up by a study using tamoxifen-induced TP53 expression on a TP53-deficient genetic background. When TP53 expression was limited to the time of irradiation, DNA damage led to strong apoptosis of hematopoietic cells and subsequently to cancer development. In contrast, when TP53 expression was only allowed at later time points, no apoptosis was induced by irradiation but also no cancer manifested. Thus, the tumor-preventive function of TP53 is not critical during the acute elimination of damaged cells but rather essential at later time points when cells that survived irradiation despite carrying genetic aberrations drive tumor progression.77
The observations in PUMA-deficient mice are consistent with these results and suggest that TP53-dependent apoptosis triggered during an acute DNA damage response is not only irrelevant for tumorigenesis, but even promotes lymphoma formation. This is underlined by the finding that resistance to radiation-induced lymphoma in PUMA-deficient mice can be overcome by PUMA-independent apoptosis induction, that is, by glucocorticoid treatment upon irradiation.72 The tumor-initiating cells in this tumor model are hematopoietic stem or progenitor cells, because T-cell specific overexpression of pro-survival Bcl-xL failed to prevent irradiation-induced lymphomagenesis despite protecting thymocytes and peripheral T cells from death whereas mice overexpressing BCL2 throughout hematopoiesis phenocopied PUMA-deficient mice.71, 72 Strikingly, in wild-type mice, persisting PUMA- and TP53-dependent apoptosis is still detected 1 week after irradiation specifically in hematopoietic progenitors as compared with more differentiated cells indicating excessive pressure within the proliferating progenitor compartment to compensate for the cell loss.78
The association between death-driven proliferation and cancer is best established in mouse models of hepatocellular carcinoma (HCC). In humans, HCC almost invariably develops in the context of chronic liver inflammation that is linked to tissue injury and cell death caused by viral hepatitis, chronic alcohol consumption, excessive hepatosteatosis or environmental toxins.79 The regenerative response is accompanied by a release of pro-inflammatory factors by dying hepatocytes and subsequent expansion of un-differentiated precursors such as tissue stem cells. Given the strong impact of apoptosis on HCC development, Qiu et al.80 investigated the role of PUMA in a mouse model of carcinogen-induced liver cancer. They found that PUMA was activated by JNK1 and critically mediated carcinogen-treatment-induced apoptosis. Importantly, PUMA deficiency decreased the multiplicity and size of emerging tumors.81 Two further studies could show that liver-specific Mcl-1 deletion induced spontaneous hepatocyte apoptosis, chronic proliferation and finally caused HCC. Noteworthy, in this mouse model, HCC developed in the absence of carcinogen treatment or detectable inflammation and hepatocytes of HCC-like lesions showed a high degree in genomic instability.82, 83
Mechanisms coupling cell death and proliferation
The discussed mouse models indicate that apoptotic cells promote cell divisions of neighboring cells, a process that can be termed death-driven proliferation, but the underlying signaling events remain elusive.84 Only recently, evidence has emerged how apoptotic cells can promote the proliferation of surrounding cells. Planarians regenerate complete individuals from the smallest of body parts upon injury85 by a process termed compensatory proliferation.86, 87 Apoptosis mediated by pro-death effectors like the caspase-like gene 3 (DjCLg3) is not restricted to the wound but occurs in primarily unaffected tissue and is thus actively involved in driving full restoration of body pieces.88 In Hydra, head regeneration after amputation requires secretion of Wnt3 by dying cells, thus initiating β-catenin-driven proliferation of surrounding cells. Blocking apoptosis by caspase inhibitors prevents head regeneration and can be overcome by exogenous Wnt3.89 In the Drosophila wing imaginal disc, a highly proliferative tissue, radiation-induced apoptosis is followed by rapid tissue regeneration to form adult structures of normal size and shape (Figure 2). Mechanistically, activity of the initiator-caspase Dronc in apoptotic cells promotes JNK and Wingless signaling pathways, thus causing the secretion of mitogens Decapentaplegic (Dpp) and Wingless (Wg) to promote tissue regeneration.90, 91, 92 Keeping apoptosis-initiated cells artificially alive by inhibiting downstream effector caspases prevented injury-induced death. Persistence of such 'undead' cells resulted in excessive proliferation and hyperplastic overgrowth due to continuous and inappropriate secretion of mitogens. In contrast to proliferating tissues where Dronc-initiated apoptosis induced Dpp and Wg expression, apoptosis-induced proliferation in committed non-dividing photoreceptor neurons in Drosophila larvae required activity of the effector caspases DrICE and Dcp-1, which subsequently force cell cycle entry mediated by Hedgehog (Hh) signaling.93
Extrapolating to human tumors, the ability of apoptotic cells to actively promote proliferation of surrounding cells, for example by secreting mitogens, might be of major significance. We speculate that cell–cell communication likely couples proliferation and cell death either passively or through signals actively elicited by apoptotic cells. In support of the latter, a recent study in xenotransplanted mice suggests that dying cells directly induce proliferation of neighboring cells. Upon radiotherapy-induced tumor cell apoptosis, caspase-3 activity led to the activation of iPLA2 and subsequent release of prostaglandin E2 by apoptotic tumor cells and neighboring stroma. Prostaglandin E2 in turn served as promoter of tumor cell survival and proliferation.94 In this model, the net response to therapy was determined by radiation-induced tumor cell apoptosis and prostaglandin E2-driven cell survival and proliferation.
An additional layer of communication between dying and proliferating cells is provided by immune and inflammatory cells. Dying cells activate macrophages, dendritic cells, neutrophils and mast cells that secrete mitogenic cytokines such as IL1, IL6 or TNFα.95 Thus, next to their major function of immune surveillance,96 the immune and inflammatory systems also foster malignant transformation under certain circumstances.95
In sum, we propose that understanding the impact of death-driven proliferation on tumorigenesis, either directly or mediated by inflammatory signals, can open a new avenue to improve therapy and potentially prevent cancer development.
From animal models to human disease
The animal models discussed earlier indicate that too much apoptosis compromises healthy or premalignant tissues by increasing proliferative pressure and clonal selection fostering outgrowth of malignant clones. But do these observations reflect tumorigenesis as it occurs in humans? And if yes, which tissues and cancer types could be affected?
The concept of apoptosis-driven cancer can be applied to therapy-induced secondary tumors that originate from distinct tissues than the primary tumors. These tumors arise as a consequence of genomic instability97 likely provoked by repeated cycles of excessive apoptosis and subsequent proliferation during therapy. Typically, they compromise tissues with high regenerative capacity (i.e., breast, intestine, rectum, skin or thyroid gland) and frequently present in the area of previous irradiation.
Adult survivors of childhood cancer have a sixfold increased risk to develop secondary tumors later in life.98 This is either due to underlying genetic (e.g., germline mutations in cancer susceptibility genes99) and/or environmental factors (e.g., nicotine abuse) that predispose these patients to tumors or to previous therapies including chemotherapy or irradiation. Patients who suffer from a combination of an underlying genetic predisposition and earlier application of chemotherapy or radiation therapy have an excessively high risk to develop secondary tumors.100
Bone marrow, a radiosensitive tissue,101 frequently gives rise to secondary malignancies as suggested by the above-described mouse model of irradiation-induced lymphomagenesis.71, 72 However, thymic lymphomas are rare in humans and do not occur as therapy-induced malignancies. In contrast, in humans, exposure to radio- or chemotherapy rather increases the risk to develop therapy-related myelodysplastic syndromes (MDS).102
MDS are clonal malignancies originating from defective bone-marrow-derived HSCs, in which critical driver mutations provide them with a selective advantage (Figure 3). This disease is characterized by ineffective hematopoiesis causing peripheral cytopenia(s) and bone marrow dysplasia. Abnormal clonal progenitor cell differentiation and increased susceptibility of immature progenitors to apoptosis underlie these symptoms. The risk to develop MDS increases with age, suggesting that accumulation of genetic damage influences pathogenesis. Exposure to alkylating agents, chemo- or radiotherapy of cancer patients dramatically increases the risk to develop therapy-related MDS (lifetime risk of 2–10%).102 MDS has a high propensity to progress to MDS-related AML (MDR-AML). Disease progression is characterized by an increased percentage of bone marrow blast cells and cytogenetic abnormalities (reviewed by Corey et al.102).

The rise and fall of apoptosis during MDS pathogenesis. Therapy-related myelodysplastic syndrom (MDS) is caused by repeated cycles of radio- or chemoradiotherapy (i.e., including alkylating agents) that lead to bone marrow attrition and subsequent regeneration. In children and adolescents, MDS can develop on the basis of congenital bone marrow failure syndrome such as Fanconi anemia and Dyskeratosis congenita. MDS frequently progresses to MDS-related AML (MDR-AML). The stepwise evolution of MDS is reflected by the FAB classification, which distinguishes between refractory anemia (RA), RA with excess blasts (RAEB), RAEB in transformation (RAEB-T) and MDR-AML
In analogy to therapy-related MDS of adults, children and adolescents can develop secondary MDS and MDR-AML on the basis of congenital bone marrow failure syndromes (Figure 3). These syndromes are caused by gene mutations affecting diverse cellular pathways but all resulting in premature failure of hematopoiesis. In individuals with these congenital conditions, HSCs become prematurely exhausted and are excessively susceptible to apoptosis or senescence.103 The most frequent bone marrow failure syndromes are Fanconi anemia, caused by mutations in DNA repair genes, and dyskeratosis congenita, characterized by premature telomere shortening.103, 104 Fanconi anemia and dyskeratosis congenita have an inherent risk to transform into MDS, with prevalence of 30–40% and 10–15%, respectively.105 Additionally, patients with these syndromes are at risk to develop other malignancies, with those children having the highest risk that previously were subjected to chemotherapeutic agents or irradiation.105
Both, therapy-related and secondary MDS are caused by cumulative HSC injury via DNA damage or oxidative stress. In patients with or animal models of bone marrow failure syndromes and low-risk MDS, HSCs are exceptionally susceptible to apoptosis.106, 107, 108 This indicates that, in line with the aforementioned animal models, excessive apoptosis generates vacant cell compartments that subsequently are repopulated by more competitive HSCs. Proliferative pressure is further increased by peripheral cytopenias and feedback loops to the bone marrow. Thus, apoptosis might be a major driver of disease progression during early stages of MDS and final transformation to full-blown AML. The pathophysiology of Fanconi anemia, dyskeratosis congenita and therapy-related MDS suggests that chronic HSC apoptosis can be attributed, at least in part, to chronic DNA damage checkpoint signaling, with ATM/ATR, CHK1, CHK2 and TP53 being central players.109 These checkpoints preserve genetic stability and act as a barrier to malignant transformation,110 thus evolving tumor cells are in need to inactivate them. Accordingly, the amount of apoptotic CD34+ HSCs gradually decreases during further progression to MDR-AML. The pressure to inactivate DNA damage checkpoint signaling is reflected by the fact that therapy-related AML more frequently harbor TP53 mutations than AML developing de novo111 and that clones harboring TP53 mutations are selected during malignant transformation of therapy-related AML.112 Similarly, CHK1 and CHK2 are strongly activated in MDS, but almost completely inactivated in MDR-AML cells.113
We have learned from animal models of bone marrow failure and MDS as well as patients’ subgroups that cells with activated checkpoint signaling display competitive disadvantages. Accordingly, checkpoint abrogation rescues proliferation and survival of HSCs, but also increases the risk of malignant transformation.109, 114, 115 What if, instead of checkpoint abrogation, apoptosis would be inhibited in early-stage MDS whereas all other pathways downstream of the DNA damage checkpoint remain active? On the basis of observations made in the murine thymic lymphoma model, we would expect an increase in bone marrow cellularity, thus relaxing proliferative pressure and delaying further transformation from MDS to MDR-AML. Indeed, the first MDS mouse model available supports this concept: Slape and colleagues116 recently showed that BCL-2 overexpression in NHD13 mice corrects macrocytic anemia and delays leukemic transformation. Further mouse models will be required to elucidate the role of apoptosis susceptibility or resistance, respectively, for pathogenesis and progression of bone marrow failure syndromes and MDS to AML.
Perspectives
Moving away from the paradigmatic view prevailing the last decades, the relationship between cell death and cancer gets far more complex than originally anticipated. Beyond doubt, the traditional view that intrinsic death of potentially dangerous cells is preventive to tumor development still holds true in many aspects. However, it disregards the fact that cells are placed in and interact with their environment. At the first glance, the dual function of apoptosis in tumorigenesis is a challenging concept, but these conflicting roles are not incompatible with common beliefs and might also depend on the tissue and the sequence of events during transformation. The balance between proliferation, senescence and death likely adapts during tumor progression. Tumor initiation must be accompanied by the odd survival of single 'initiated' cells that carry driver mutations. Recently, light has been shed on the process of initiation of hematological malignancies. Driver mutations conferring HSCs with selective advantages (i.e., in DNMT3A, JAK2, ASXL1, TET2 and others) lead to clonal expansion in aged individuals, who do not (yet) suffer from leukemia or MDS.117 During further tumor progression, cancer cells frequently respond to their altered state by undergoing programmed cell death and remain highly dependent on certain survival signals from their environment.118, 119 Within a growing tumor, apoptosis will preferentially eliminate those sub-clones with the highest apoptosis sensitivity whereas sparing the more resistant cells. Thus, cell death imposes a huge selection pressure favoring clonal expansion of more aggressive sub-clones. Hence, even fully established tumors are rarely completely resistant to apoptosis, and death induced by hypoxia or chemotherapeutics increases proliferative pressure and clonal selection paving the way for therapy-refractory or relapsing cancers.
In light of these findings, it becomes apparent that standard anti-cancer therapies face a dilemma by aiming at inducing tumor cell death. Hence, we believe that we are in need for better treatment strategies to avoid unnecessary tissue damage and inflammatory responses in tumor patients as well as in patients presenting with premalignant conditions such as bone marrow failure or viral hepatitis. We are still only beginning to understand the complex mechanisms involved in tumor development and progression, and thus further research is necessary to understand the contribution of apoptosis in shaping tumors, as a prerequisite to generate a more comprehensive picture on tumorigenesis and allow more effective therapeutic intervention.
Abbreviations
- BH3-only:
-
BCL-2 homology domain 3-only
- HCC:
-
hepatocellular carcinoma
- HSC:
-
hematopoietic stem cell
- MDS:
-
myelodysplastic syndrome
- MDR-AML:
-
MDS-related acute myeloid leukemia
- RA:
-
refractory anemia
- RAEB:
-
RA with excess blasts
- RAEB-T:
-
RAEB in transformation
References
Harris CC . Chemical and physical carcinogenesis: advances and perspectives for the 1990s. Cancer Res 1991; 51: 5023s–5044s.
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Green DR, Evan GI . A matter of life and death. Cancer Cell 2002; 1: 19–30.
Lowe SW, Lin AW . Apoptosis in cancer. Carcinogenesis 2000; 21: 485–495.
Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 1985; 41: 899–906.
Cleary ML, Sklar J . Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A 1985; 82: 7439–7443.
Tsujimoto Y, Cossman J, Jaffe E, Croce CM . Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985; 228: 1440–1443.
Amundson SA, Myers TG, Fornace AJ Jr . Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene 1998; 17: 3287–3299.
Levine AJ . p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331.
Rufini A, Tucci P, Celardo I, Melino G . Senescence and aging: the critical roles of p53. Oncogene 2013; 32: 5129–5143.
Michalak E, Villunger A, Erlacher M, Strasser A . Death squads enlisted by the tumour suppressor p53. Biochem Biophys Res Commun 2005; 331: 786–798.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003; 302: 1036–1038.
Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L et al. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood 2005; 106: 4131–4138.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288: 1053–1058.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003; 4: 321–328.
Ichikawa A, Kinoshita T, Watanabe T, Kato H, Nagai H, Tsushita K et al. Mutations of the p53 gene as a prognostic factor in aggressive B-cell lymphoma. N Engl J Med 1997; 337: 529–534.
Moller MB, Gerdes AM, Skjodt K, Mortensen LS, Pedersen NT . Disrupted p53 function as predictor of treatment failure and poor prognosis in B- and T-cell non-Hodgkin's lymphoma. Clin Cancer Res 1999; 5: 1085–1091.
Sherr CJ, Weber JD . The ARF/p53 pathway. Curr Opin Genet Dev 2000; 10: 94–99.
Khanna KK, Jackson SP . DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 2001; 27: 247–254.
Schubbert S, Shannon K, Bollag G . Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7: 295–308.
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G . Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997; 278: 687–689.
Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 2003; 22: 6785–6793.
Wu DW, Huang CC, Chang SW, Chen TH, Lee H . Bcl-2 stabilization by paxillin confers 5-fluorouracil resistance in colorectal cancer. Cell Death Differ e-pub ahead of print 17 October 2014.
Nesbit CE, Tersak JM, Prochownik EV . MYC oncogenes and human neoplastic disease. Oncogene 1999; 18: 3004–3016.
Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009; 113: 4403–4413.
Mackus WJ, Kater AP, Grummels A, Evers LM, Hooijbrink B, Kramer MH et al. Chronic lymphocytic leukemia cells display p53-dependent drug-induced Puma upregulation. Leukemia 2005; 19: 427–434.
Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, Osorio LM . Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia 2004; 18: 41–47.
Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A . Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 2007; 117: 112–121.
Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood 1998; 91: 3379–3389.
Hamilton E, Pearce L, Morgan L, Robinson S, Ware V, Brennan P et al. Mimicking the tumour microenvironment: three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br J Haematol 2012; 158: 589–599.
Asslaber D, Grossinger EM, Girbl T, Hofbauer SW, Egle A, Weiss L et al. Mimicking the microenvironment in chronic lymphocytic leukaemia - where does the journey go? Br J Haematol 2013; 160: 711–714.
Llambi F, Green DR . Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev 2011; 21: 12–20.
Kaspers GJ, Pieters R, Van Zantwijk CH, Van Wering ER, Van Der Does-Van Den Berg A, Veerman AJ . Prednisolone resistance in childhood acute lymphoblastic leukemia: vitro-vivo correlations and cross-resistance to other drugs. Blood 1998; 92: 259–266.
Ploner C, Rainer J, Niederegger H, Eduardoff M, Villunger A, Geley S et al. The BCL2 rheostat in glucocorticoid-induced apoptosis of acute lymphoblastic leukemia. Leukemia 2008; 22: 370–377.
Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci USA 2006; 103: 14907–14912.
Calandra G, Bridger G, Fricker S . CXCR4 in clinical hematology. Curr Top Microbiol Immunol 2010; 341: 173–191.
Labi V, Grespi F, Baumgartner F, Villunger A . Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ 2008; 15: 977–987.
Cragg MS, Harris C, Strasser A, Scott CL . Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 2009; 9: 321–326.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68: 3421–3428.
Mazumder S, Choudhary GS, Al Harbi S, Almasan A . Mcl-1 Phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res 2012; 72: 3069–3079.
Rhiner C, Moreno E . Super competition as a possible mechanism to pioneer precancerous fields. Carcinogenesis 2009; 30: 723–728.
Okada H, Mak TW . Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 2004; 4: 592–603.
Dahan P, Martinez GJ, Delmas C, Monferran S, Malric L, Zentkowski D et al. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: possible involvement in radioresistance. Cell Death Dis 2014; 5: e1543.
Cotter TG . Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 2009; 9: 501–507.
Strasser A, Harris AW, Bath ML, Cory S . Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990; 348: 331–333.
Roulland S, Kelly RS, Morgado E, Sungalee S, Solal-Celigny P, Colombat P et al. t(14;18) Translocation: A predictive blood biomarker for follicular lymphoma. J Clin Oncol 2014; 32: 1347–1355.
Vaux DL, Cory S, Adams JM . Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988; 335: 440–442.
Fanidi A, Harrington EA, Evan GI . Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature 1992; 359: 554–556.
Beverly LJ, Varmus HE . MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 2009; 28: 1274–1279.
Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 2014; 28: 58–70.
Frenzel A, Labi V, Chmelewskij W, Ploner C, Geley S, Fiegl H et al. Suppression of B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood 2010; 115: 995–1005.
Egle A, Harris AW, Bouillet P, Cory S . Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA 2004; 101: 6164–6169.
Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ 2009; 16: 684–696.
Jager R, Herzer U, Schenkel J, Weiher H . Overexpression of Bcl-2 inhibits alveolar cell apoptosis during involution and accelerates c-myc-induced tumorigenesis of the mammary gland in transgenic mice. Oncogene 1997; 15: 1787–1795.
Jager R . Targeting the death machinery in mammary epithelial cells: Implications for breast cancer from transgenic and tissue culture experiments. Crit Rev Oncol Hematol 2007; 63: 231–240.
Evan G, Littlewood T . A matter of life and cell death. Science 1998; 281: 1317–1322.
Hogarth LA, Hall AG . Increased BAX expression is associated with an increased risk of relapse in childhood acute lymphocytic leukemia. Blood 1999; 93: 2671–2678.
Gurova KV, Kwek SS, Koman IE, Komarov AP, Kandel E, Nikiforov MA et al. Apoptosis inhibitor as a suppressor of tumor progression: expression of Bcl-2 eliminates selective advantages for p53-deficient cells in the tumor. Cancer Biol Ther 2002; 1: 39–44.
Luke JJ, Van De Wetering CI, Knudson CM . Lymphoma development in Bax transgenic mice is inhibited by Bcl-2 and associated with chromosomal instability. Cell Death Differ 2003; 10: 740–748.
Moreno E, Basler K . dMyc transforms cells into super-competitors. Cell 2004; 117: 117–129.
Li W, Baker NE . Engulfment is required for cell competition. Cell 2007; 129: 1215–1225.
Meyer SN, Amoyel M, Bergantinos C, de la CC, Schertel C, Basler K et al. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science 2014; 346: 1258236.
Morata G, Ballesteros-Arias L . Developmental Biology Death to the losers. Science 2014; 346: 1181–1182.
Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature 2014; 509: 465–470.
Moreno E . Cancer: Darwinian tumour suppression. Nature 2014; 509: 435–436.
Dewanji A, Luebeck EG, Moolgavkar SH . A generalized Luria-Delbruck model. Math Biosci 2005; 197: 140–152.
Komarova NL, Wodarz D . Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci U S A 2005; 102: 9714–9719.
Enderling H, Anderson AR, Chaplain MA, Beheshti A, Hlatky L, Hahnfeldt P . Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res 2009; 69: 8814–8821.
Enderling H, Hahnfeldt P . Cancer stem cells in solid tumors: is 'evading apoptosis' a hallmark of cancer? Prog Biophys Mol Biol 2011; 106: 391–399.
Wodarz D, Komarova N . Can loss of apoptosis protect against cancer? Trends Genet 2007; 23: 232–237.
Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J et al. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev 2010; 24: 1602–1607.
Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM et al. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev 2010; 24: 1608–1613.
Kemp CJ, Wheldon T, Balmain A . p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet 1994; 8: 66–69.
Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol 2008; 28: 5391–5402.
Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J . Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol 2010; 8: e1000324.
Bondar T, Medzhitov R . p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010; 6: 309–322.
Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI . The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006; 443: 214–217.
Labi V, Villunger A . PUMA-mediated tumor suppression: A tale of two stories. Cell Cycle 2010; 9: 4269–4275.
El Serag HB, Rudolph KL . Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132: 2557–2576.
Qiu W, Wu B, Wang X, Buchanan ME, Regueiro MD, Hartman DJ et al. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest 2011; 121: 1722–1732.
Baumgartner F, Villunger A . Apoptosis: a barrier against cancer no more? Hepatology 2011; 54: 1121–1124.
Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010; 51: 1226–1236.
Vick B, Weber A, Urbanik T, Maass T, Teufel A, Krammer PH et al. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology 2009; 49: 627–636.
Mollereau B, Perez-Garijo A, Bergmann A, Miura M, Gerlitz O, Ryoo HD et al. Compensatory proliferation and apoptosis-induced proliferation: a need for clarification. Cell Death Differ 2013; 20: 181.
Gurtner GC, Werner S, Barrandon Y, Longaker MT . Wound repair and regeneration. Nature 2008; 453: 314–321.
Bergmann A, Steller H . Apoptosis, stem cells, and tissue regeneration. Sci Signal 2010: 3re8.
Fan Y, Bergmann A . Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell!. Trends Cell Biol 2008; 18: 467–473.
Hwang JS, Kobayashi C, Agata K, Ikeo K, Gojobori T . Detection of apoptosis during planarian regeneration by the expression of apoptosis-related genes and TUNEL assay. Gene 2004; 333: 15–25.
Chera S, Ghila L, Dobretz K, Wenger Y, Bauer C, Buzgariu W et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell 2009; 17: 279–289.
Perez-Garijo A, Martin FA, Morata G . Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 2004; 131: 5591–5598.
Ryoo HD, Gorenc T, Steller H . Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 2004; 7: 491–501.
Huh JR, Guo M, Hay BA . Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol 2004; 14: 1262–1266.
Fan Y, Bergmann A . Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell 2008; 14: 399–410.
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 2011; 17: 860–866.
Mantovani A, Allavena P, Sica A, Balkwill F . Cancer-related inflammation. Nature 2008; 454: 436–444.
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515: 577–581.
Sigurdson AJ, Jones IM . Second cancers after radiotherapy: any evidence for radiation-induced genomic instability? Oncogene 2003; 22: 7018–7027.
Landier W, Bhatia S . Cancer survivorship: a pediatric perspective. Oncologist 2008; 13: 1181–1192.
Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH . Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990; 348: 747–749.
Wong JR, Morton LM, Tucker MA, Abramson DH, Seddon JM, Sampson JN et al. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol 2014; 32: 3284–3290.
Ringshausen I, O'Shea CC, Finch AJ, Swigart LB, Evan GI . Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10: 501–514.
Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD . Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer 2007; 7: 118–129.
Alter BP . Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program 2007: 29–39.
Soulier J . Fanconi anemia. Hematology Am Soc Hematol Educ Program 2011; 2011: 492–497.
Alter BP, Giri N, Savage SA, Peters JA, Loud JT, Leathwood L et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 2010; 150: 179–188.
Shimazaki K, Ohshima K, Suzumiya J, Kawasaki C, Kikuchi M . Apoptosis and prognostic factors in myelodysplastic syndromes. Leuk Lymphoma 2002; 43: 257–260.
Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A . The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000; 96: 3932–3938.
Tehranchi R, Fadeel B, Forsblom AM, Christensson B, Samuelsson J, Zhivotovsky B et al. Granulocyte colony-stimulating factor inhibits spontaneous cytochrome c release and mitochondria-dependent apoptosis of myelodysplastic syndrome hematopoietic progenitors. Blood 2003; 101: 1080–1086.
Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012; 11: 36–49.
Zhou BB, Elledge SJ . The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433–439.
Side LE, Curtiss NP, Teel K, Kratz C, Wang PW, Larson RA et al. RAS, FLT3, and TP53 mutations in therapy-related myeloid malignancies with abnormalities of chromosomes 5 and 7. Genes Chromosomes Cancer 2004; 39: 217–223.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature e-pub ahead of print 8 December 2014.
Boehrer S, Ades L, Tajeddine N, Hofmann WK, Kriener S, Bug G et al. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene 2009; 28: 2205–2218.
Ceccaldi R, Briot D, Larghero J, Vasquez N, Dubois dC, Chamousset D . Spontaneous abrogation of the G(2)DNA damage checkpoint has clinical benefits but promotes leukemogenesis in Fanconi anemia patients. J Clin Invest 2011; 121: 184–194.
Waisfisz Q, Morgan NV, Savino M, de Winter JP, van Berkel CG, Hoatlin ME . Spontaneous functional correction of homozygous fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet 1999; 22: 379–383.
Slape CI, Saw J, Jowett JB, Aplan PD, Strasser A, Jane SM et al. Inhibition of apoptosis by BCL2 prevents leukemic transformation of a murine myelodysplastic syndrome. Blood 2012; 120: 2475–2483.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20: 1472–1478.
Jaattela M . Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene 2004; 23: 2746–2756.
Kroemer G, Martin SJ . Caspase-independent cell death. Nat Med 2005; 11: 725–730.
Labi V, Erlacher M, Kiessling S, Villunger A . BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ 2006; 13: 1325–1338.
Buglioni S, D'Agnano I, Cosimelli M, Vasselli S, D'Angelo C, Tedesco M et al. Evaluation of multiple bio-pathological factors in colorectal adenocarcinomas: independent prognostic role of p53 and bcl-2. Int J Cancer 1999; 84: 545–552.
Kaklamanis L, Savage A, Whitehouse R, Doussis-Anagnostopoulou I, Biddolph S, Tsiotos P et al. Bcl-2 protein expression: association with p53 and prognosis in colorectal cancer. Br J Cancer 1998; 77: 1864–1869.
Manne U, Myers RB, Moron C, Poczatek RB, Dillard S, Weiss H et al. Prognostic significance of Bcl-2 expression and p53 nuclear accumulation in colorectal adenocarcinoma. Int J Cancer 1997; 74: 346–358.
Meterissian SH, Kontogiannea M, Al Sowaidi M, Linjawi A, Halwani F, Jamison B et al. Bcl-2 is a useful prognostic marker in Dukes' B colon cancer. Ann Surg Oncol 2001; 8: 533–537.
Ofner D, Riehemann K, Maier H, Riedmann B, Nehoda H, Totsch M et al. Immunohistochemically detectable bcl-2 expression in colorectal carcinoma: correlation with tumour stage and patient survival. Br J Cancer 1995; 72: 981–985.
Watson NF, Madjd Z, Scrimegour D, Spendlove I, Ellis IO, Scholefield JH et al. Evidence that the p53 negative / Bcl-2 positive phenotype is an independent indicator of good prognosis in colorectal cancer: a tissue microarray study of 460 patients. World J Surg Oncol 2005; 3: 47.
Berardo MD, Elledge RM, de Moor C, Clark GM, Osborne CK, Allred DC . Bcl-2 and apoptosis in lymph node positive breast carcinoma. Cancer 1998; 1296: 82.
Binder C, Marx D, Overhoff R, Binder L, Schauer A, Hiddemann W . Bcl-2 protein expression in breast cancer in relation to established prognostic factors and other clinicopathological variables. Ann Oncol 1995; 6: 1005–1010.
Castiglione F, Sarotto I, Fontana V, Destefanis M, Venturino A, Ferro S et al. Bcl2, p53 and clinical outcome in a series of 138 operable breast cancer patients. Anticancer Res 1999; 19: 4555–4563.
Daidone MG, Veneroni S, Benini E, Tomasic G, Coradini D, Mastore M et al. Biological markers as indicators of response to primary and adjuvant chemotherapy in breast cancer. Int J Cancer 1999; 84: 580–586.
Dawson SJ, Makretsov N, Blows FM, Driver KE, Provenzano E, Le Quesne J et al. BCL2 in breast cancer: a favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br J Cancer 2010; 103: 668–675.
Friedrich K, Dimmer V, Haroske G, Lossnitzer A, Kasper M, Theissig F et al. Expression of p53 and bcl-2 in correlation to clinicopathological parameters, hormone receptor status and DNA ploidy in breast cancers. Pathol Res Pract 1995; 191: 1114–1121.
Joensuu H, Pylkkanen L, Toikkanen S . Bcl-2 protein expression and long-term survival in breast cancer. Am J Pathol 1994; 145: 1191–1198.
Kobayashi S, Iwase H, Ito Y, Yamashita H, Iwata H, Yamashita T et al. Clinical significance of bcl-2 gene expression in human breast cancer tissues. Breast Cancer Res Treat 1997; 42: 173–181.
Silvestrini R, Veneroni S, Daidone MG, Benini E, Boracchi P, Mezzetti M et al. The Bcl-2 protein: a prognostic indicator strongly related to p53 protein in lymph node-negative breast cancer patients. J Natl Cancer Inst 1994; 86: 499–504.
Vargas-Roig LM, Cuello-Carrion FD, Fernandez-Escobar N, Daguerre P, Leuzzi M, Ibarra J et al. Prognostic value of Bcl-2 in breast cancer patients treated with neoadjuvant anthracycline based chemotherapy. Mol Oncol 2008; 2: 102–111.
Villar E, Redondo M, Rodrigo I, Garcia J, Avila E, Matilla A . Bcl-2 expression and apoptosis in primary and metastatic breast carcinomas. Tumour Biol 2001; 22: 137–145.
Zhang GJ, Kimijima I, Abe R, Watanabe T, Kanno M, Hara K et al. Apoptotic index correlates to bcl-2 and p53 protein expression, histological grade and prognosis in invasive breast cancers. Anticancer Res 1998; 18: 1989–1998.
Anagnostou VK, Lowery FJ, Zolota V, Tzelepi V, Gopinath A, Liceaga C et al. High expression of BCL-2 predicts favorable outcome in non-small cell lung cancer patients with non squamous histology. BMC Cancer 2010; 10: 186.
Renouf DJ, Wood-Baker R, Ionescu DN, Leung S, Masoudi H, Gilks CB et al. BCL-2 expression is prognostic for improved survival in non-small cell lung cancer. J Thorac Oncol 2009; 4: 486–491.
Shibata Y, Hidaka S, Tagawa Y, Nagayasu T . Bcl-2 protein expression correlates with better prognosis in patients with advanced non-small cell lung cancer. Anticancer Res 2004; 24: 1925–1928.
Zhao XD, He YY, Gao J, Zhao C, Zhang LL, Tian JY et al. High expression of Bcl-2 protein predicts favorable outcome in non-small cell lung cancer: evidence from a systematic review and meta-analysis. Asian Pac J Cancer Prev 2014; 15: 8861–8869.
Pillai K, Pourgholami MH, Chua TC, Morris DL . Does the expression of BCL2 have prognostic significance in malignant peritoneal mesothelioma? Am J Cancer Res 2013; 3: 312–322.
McDonald FE, Ironside JW, Gregor A, Wyatt B, Stewart M, Rye R . The prognostic influence of bcl-2 in malignant glioma. Br J Cancer 2002; 86: 1899–1904.
Inada T, Kikuyama S, Ichikawa A, Igarashi S, Ogata Y . Bcl-2 expression as a prognostic factor of survival of gastric carcinoma. Anticancer Res 1998; 18: 2003–2010.
Acknowledgements
We thank Andreas Villunger, Emmanuel Derudder and Alexander Egle for insightful discussions. VL is supported by an EMBO fellowship (ALTF 1146-2010) and by the TWF (Tiroler Wissenschaftsfonds UNI-0404/1696). ME is funded by the German Research Foundation (FOR 2036) and the Sander-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International Licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0
About this article

Cite this article
Labi, V., Erlacher, M. How cell death shapes cancer. Cell Death Dis 6, e1675 (2015). https://doi.org/10.1038/cddis.2015.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.20
This article is cited by
-
Molecularly imprinted polymers (MIPs): emerging biomaterials for cancer theragnostic applications
Biomaterials Research (2023)
-
Tumor-targeted superantigens produce curative tumor immunity with induction of memory and demonstrated antigen spreading
Journal of Translational Medicine (2023)
-
Hypomethylation-mediated upregulation of the WASF2 promoter region correlates with poor clinical outcomes in hepatocellular carcinoma
Journal of Experimental & Clinical Cancer Research (2022)
-
Spontaneous activity of the mitochondrial apoptosis pathway drives chromosomal defects, the appearance of micronuclei and cancer metastasis through the Caspase-Activated DNAse
Cell Death & Disease (2022)
-
Tannic acid enhances cisplatin effect on cell proliferation and apoptosis of human osteosarcoma cell line (U2OS)
Pharmacological Reports (2022)
