
Abstract
The short isoform of ErbB3-binding protein 1 (Ebp1), p42, is considered to be a potent tumor suppressor in a number of human cancers, although the mechanism by which it exerts this tumor-suppressive activity is unclear. Here, we report that p42 interacts with the cSH2 domain of the p85 subunit of phosphathidyl inositol 3-kinase (PI3K), leading to inhibition of its lipid kinase activity. Importantly, we found that p42 induces protein degradation of the p85 subunit and further identified HSP70/CHIP complex as a novel E3 ligase for p85 that is responsible for p85 ubiquitination and degradation. In this process, p42 couples p85 to the HSP70/CHIP-mediated ubiquitin–proteasomal system (UPS), thereby promoting a reduction of p85 levels both in vitro and in vivo. Thus, the tumor-suppressing effects of p42 in cancer cells are driven by negative regulation of the p85 subunit of PI3K.
Similar content being viewed by others


Main
ErbB3-binding protein 1 (Ebp1) has been implicated in a number of human cancers. It has two isoforms, p48 and p42, that have different cellular activities.1, 2 In mammalian cells, p48 is the predominant protein whereas p42 is selectively degraded through ubiquitination.2, 3 The crystal structure of Ebp1 isoforms shows that p42 is missing one and a half helices at the amino-terminus,4 implying that conformational changes associated with 54 fewer amino acid residues may account for the possible differential roles of Ebp1 isoforms. Recently, we showed that p48 acts as an oncogene by promoting tumorigenicity of glioma cells through the downregulation of p53. In contrast, p42 is considered to be a potent tumor suppressor because of its inhibitory growth function.5, 6 Numerous p42-binding partners have been identified, but none have provided substantial insight into the tumor-suppressor activity of p42. Despite growing interest in the differential role of Ebp1 isoforms in tumorigenesis and the newly defined molecular mechanisms of p48 isoform contributions to cancer cell growth, the molecular mechanism accounting for the tumor-suppressor activity of short isoform p42 remains unknown.
The aberrant activation of the phosphathidyl inositol 3-kinase (PI3K)/Akt signaling pathway is commonly observed in many cancers. Among three classes of PI3Ks, Class IA PI3Ks (hereafter referred to generically PI3K) are the most defined and the only class clearly implicated in human cancers.7 PI3K is composed of a p110 catalytic subunit and p85 regulatory subunit. Regulatory subunits have multiple roles in the function of PI3K such as downregulation of the basal activity, stabilization of the catalytic subunit and sequential activation by tyrosine receptor kinases and Ras.8, 9, 10 The representative regulatory subunit, p85α, contains several modular protein–protein interaction domain; a Src-homology 3 (SH3) domain, a breakpoint clustered homology (BH) domain, two Src homology 2 (SH2) domains and an inter-SH2 (iSH2) domain. The iSH2 domain is the primary p110α-binding domain.11, 12 The nSH2 and cSH2 domains bind phosphorylated tyrosine in activated receptors and adaptor proteins, and this interaction relieves the basal inhibition of p110 subunit, allowing for the activation of the catalytic subunit. Because most cancer-associated mutations have been reported in p110, p110 is considered as an attractive therapeutic target. However, oncogenic mutations in p85 have also been found in the iSH2 domain as a truncation, deletion or point mutation, presumably by disrupting inhibitory contacts with p110 or failing to inhibit p110. Thus, studying regulation of p85 may provide novel strategy for cancer treatment.
Ubiquitin–proteasome system (UPS) is the main pathway for eliminating misfiled proteins,13, 14 and molecular chaperones assist protein folding but can also facilitate the ubiquitin degradation of misfolded protein15 by binding to co-chaperones. The identification of co-chaperone, CHIP (carboxy terminus of heat shock protein 70 (Hsp70)-interacting protein), has revealed the physical link from molecular chaperone to UPS by targeting chaperone substrates for proteasome degradation.16, 17 CHIP has a Ubox-dependent E3 ligase and possesses three tandem tetratricopeptide repeat motifs that provide interface with chaperones HSP70 and HSP90, and then ubiquitinates chaperone-bound substrates.18, 19 Many of oncoproteins including receptor tyrosine kinase ErbB2 and hypoxia-inducible factor-1α have been reported as a target of CHIP.
Here we show that p42 specifically interacts with p85 regulatory domain of PI3K and inhibits PI3K activity. Importantly, we find that overexpression of p42 dramatically reduced p85 protein levels by linking p85 to HSP70/CHIP-mediated proteasomal degradation. Thus, p42 functions as an important tumor suppressor in human cancer cells through negative regulation of the stability of p85 subunit. Our data provide a new insight into the mechanism of deregulation of PI3K in tumor cells, reporting HSP70/CHIP as a novel E3 ubiquitin ligase for p85 subunit.
Results
The p42 specifically interacts with p85 regulatory subunit of PI3K
Tumor suppressors normally control cell growth by mediating mitogenic signaling,20 and our studies have shown that overexpression of p42 prohibits Akt activation, whereas p48 enhances Akt kinase activity.2, 21 In an effort to determine how p42 functions as a growth suppressor, we discovered that p42 but not p48 interacts with both the endogenous and transfected p85 regulatory subunits of PI3K (Figures 1a and b). Mapping analysis showed that the N-terminal domain of Ebp1 including amino acids 1–54, which are only present in p48, is dispensable for p85 binding, and fragment 183–394 is critical for the interaction between p42 and p85 subunits (Figures 1c and d). Reciprocal experiments with various deletion mutants of p85 demonstrated that the c-SH2 domain of p85, which is responsible for binding to receptor tyrosine kinase,22 is crucial for the association with p42, but deletion of the internal SH2 domain (iSH; known as the p110 binding domain)23 had no effect on p42 binding (Figure 1e). Although we also found that p42 interacts with the p110 catalytic domain of PI3K (Supplementary Figure 1a), this interaction only occurs with the adaptor binding domain (ABD) of p110 (Supplementary Figures 1b and c), which is necessary and sufficient to bind the p85 regulatory subunit,22 implicating that the interaction between p42 and p110 is a consequence of p42–p85 complex formation (Supplementary Figure 1d).

The p42 specifically interacts with p85 regulatory subunit of PI3K. (a) HEK 293T cells were transfected as indicated. Cell lysates were immunoprecipitated with anti-Myc antibody, and endogenous p85 protein was determined by immunoblotting with the anti-p85 antibody. (b) Transfected cells were subjected to immunoprecipitation and immunoblotting as indicated (left). To detect endogenous interaction between p85 and p42 or p48, 293T cell lysates were immunoprecipitated with anti-p85 antibody and immunoblotting with anti-Ebp1 or anti-N-Ebp1 antibody (right). (c) Schematic diagram of the Ebp1 fragments. (d) Flag-p85 and GFP-Ebp1 co-transfected cells were immunoprecipitated with anti-Flag and immunoblotted as indicated. (e) The schematic diagram of various domains of the p85 subunit (left), and binding analysis between GFP-p42 and Flag-p85 fragments (right)
P42 inhibits the activity of PI3K
The ability of p42 to bind to p85 raised the question of whether this interaction influences the lipid kinase activity of PI3K in tumor cells. To test this hypothesis, we transfected GFP-tagged human p48, p42 or an empty vector control into U251 MG glioblastoma cell lines and selected stable cell clone (Supplementary Figures 2a and b). Our in vitro thin layer chromatography (TLC) showed a robust decrease in PI3K activity following increased expression of p42, whereas p48 expression did not exert any notable changes on PI3K activity, suggesting that p42 inhibits the capacity of PI3K to phosphorylate phosphatidylinositol (Figure 2a). Consistently, overexpression of p42 notably decreased the amount of final product of PI3K, PI(3,4,5)P3(PIP3) (Figure 2b), and subsequently suppressed tumor cell growth, invasion and anchorage-independent growth in soft agar with smaller and twice as less colonies in p42-expressing cells than in the vector alone or in p48-expressing cells, whereas p48 increased cell growth, invasion and soft agar growth, fitting with our previous finding (Figures 2c–e).24 In contrast, silencing of p42 enhanced lipid kinase activity relative to the control, whereas specific depletion of p48 by N-si-p48(ref.24) did not affect PI3K activity, implying that the decrease of PIP3 production upon inhibition of PI3K occurs by selective expression of p42 (Figure 2f).

The p42 controls PI3K activity. (a) Cell lysates of p42 (1 and 3 μg) or p48 (3 μg) overexpressing cells were immunoprecipitated with anti-p85 antibody and labeled with 32P. Lipids were then extracted and analyzed for TLC. **P<0.005. (b) Cells expressing the indicated constructs were applied for the detection of PIP3 levels as described in the Materials and Methods. (c) U251 glioma cells expressing the indicated constructs were plated (5 × 104 cells/60 mm dish) in complete medium and the viable cells were counted as indicated times. (d) An invasion assay was performed by plating the glioma cells expressing the indicated constructs onto matrigel-coated invasion chambers. Invasive cells were fixed and stained, and representative areas were photographed. Invasive cells were counted at × 100 magnification. (e) Representative digital microscopic images of colony-forming cells were assessed. Cells were fixed and stained with 0.1% crystal violet. Scale bar=500 μm (left upper), 100 μm (left bottom). In soft agar assays, cells stably expressing p48 and p42 were seeded on 0.35% soft agar (supplemented with complete medium) and cultured for 4 weeks (right). The colonies were counted at a magnification of × 40. Each value represents the mean±S.E.M. of triplicate measurements. *P<0.05 versus pEGFPC2 control. (f) SCR-, N-si-p48- or si-Ebp1-transfected cells were used in the PI3K activity assay
P42 controls p85 protein stability through ubiquitin–proteasome pathway
How might p42 facilitate the inhibition of PI3K activity? PI3K inhibition is a major pharmacological target for drug discovery. The primary function of the p85 subunit of PI3K is to bind, stabilize and inhibit the p110 subunit until RTK activation. Thus, to determine the molecular basis of p42-mediated inhibition of PI3K signaling, we first tested whether p42 interrupts the heterodimerization of p85 and p110. However, the interaction between p85 and p110 is little decreased upon increased p42 expression (Figure 3a) that suggests that p42 does not compete with the p110 subunit for p85 binding, and a reduction of PI3K activity by p42 might not be caused by the direct abrogation of the heterodimer. Next, we examined the influence of p42 expression on p85 membrane recruitment, an essential process of PI3K activation. Our immunostaining and fractionation analyses showed that forced expression of p42 appears to diminish membrane localization of p85 (Figures 3b and c). However, more importantly, p42-expressing cells displayed a much lower intensity of p85 fluorescence, and not only the membrane fraction, but also the cytoplasmic fraction of p85 concurrently decreased, suggesting that decreased membrane localization of p85 might be a consequence of altered p85 protein level.

The p42 regulates p85 protein stability. (a) HEK293T cells were co-transfected with GST-p110 (2 μg) and Flag-p85 (2 μg) along with different concentrations of Myc-p42 (2 or 4 μg). The cell lysates (1 mg) were subjected to a GST pull-down assay, and the interaction between p85 and p110 was shown by immunoblotting with anti-Flag antibody. Protein expression was evaluated by immunoblotting. (b) Cells from the breast cancer cell line MCF7 were transfected with GFP-p85 (2 μg) and RED-plasma membrane marker (1.5 μg) together with control (2 μg), Myc-p42 (2 μg) or Myc-p48 (2 μg). The serum-starved MCF7 cells were incubated with or without EGF (50 ng/ml) for 5 min before processing for immunocytochemistry. Cells immunostained with anti-Myc were immunoreacted with Alexa Fluor 350 donkey anti-mouse secondary antibody and observed using a Zeiss confocal laser-scanning microscope. p48 or p42 (gray), plasma membrane (red) and p85 (green). Scale bar: 10 μm. (c) Cells were transfected with Flag-p85 along with control or GFP-p42 (2 or 4 μg). The serum-starved cells were treated with EGF (50 ng/ml) for 5 min. The cell lysates were subjected to subcellular fractionation for membrane and cytosolic fraction. p85 protein levels were evaluated by immunoblotting with anti-Flag antibody. The purity of each fraction was examined by anti-tubulin (cytosol) and anti-Na/K ATPas (membrane) antibodies. The cytosolic fraction was shown in upper and the membrane fraction was shown in bottom. (d) p85 protein levels were determined from control or Myc-p42 (2(+) and 4(++) μg)-transfected HEK 293 cells by immunoblotting (upper), and quantification is shown as a bar graph (bottom). (e) Transfected PC12 cells were stained with an anti-p85 and Alexa Flour 594 goat anti-mouse antibody (gray) and visualized using a confocal microscope LSM 710 (Zeiss). The nucleus was counterstained with Hoechst (blue). Scale bar: 10 μm. (f) HEK 293T cells were transfected with different concentrations of Myc-p42 (2 or 4 μg); after 48 h, RT-PCR was performed with the extracted total RNA using p85-specific primers. (g) Cells transfected with control or GFP-p42 were incubated with cycloheximide (CHX; 10 μg/ml) for the indicated times. p85 protein levels were analyzed by immunoblotting (upper). Quantification of p85 protein levels in control (square) and GFP-p42 (circle) cells are shown (bottom); values are expressed relative to time 0 (normalized to actin). (h) HEK 293T cells transfected with SCR, N-si-p48 or si-Ebp1 were incubated with CHX (10 μg /ml) for the indicated times. p85 protein levels were analyzed by immunoblotting. (i) Control, myc-p42- or p48-transfected cells were used to determine protein levels of p85 and p110 by immunoblotting (left). Densitometry analysis (right). *P<0.05 versus control. (j) Endogenous p85 levels were determined from PC12 cells silenced with SCR, N-si-p48 or si-Ebp1 (left). Densitometry analysis (right). **P<0.005 versus control
Importantly, we found that increased p42 expression dramatically diminished p85 protein level in HEK293 (Figures 3d and e) but not RNA levels (Figure 3f), implying that p42 mediates reduction of p85 in post-translational events. Indeed, the half-life of p85 was markedly decreased in p42-expressing cells, but not in p48-expressing cells, compared with control cells, and this indicates that p85 levels were not altered by treatment with eukaryotic protein synthesis inhibitor, cycloheximide (CHX), for up to 8 h, consistent with previous reports that p85 is a relatively stable protein (Figure 3g and Supplementary Figure 3a).25 Depletion of p42 reversed this effect whereas depletion of p48 has no effect on the half-life of p85 (Figure 3h).
To ascertain that p42 mediates the specific degradation of p85 subunit in cells, we determined endogenous protein levels of both p85 and p110 subunits of PI3K. Ectopic expression of p42 remarkably diminished endogenous p85 levels, although protein levels of p110 appear to be slightly decreased (Figure 3i), and depletion of p42 restored p85 protein level, whereas depletion of p48 had no effect on p85 level (Figure 3j), suggesting that p42 probably inhibits PI3K activity by controlling p85 protein stability. Nevertheless, either p42 or p48 expression did not alter the half-life of p110 subunit (Supplementary Figure 3b).
To explore the molecular mechanisms underlying p42-induced p85 instability, we examined whether reduction of p85 is involved in proteasomal degradation. Pretreatment of MG132, a proteasomal inhibitor, efficiently protected p85 from p42-mediated degradation (Figure 4a), implying that p42 downregulates p85 protein by promoting ubiquitination-dependent proteasomal degradation. Moreover, p85 was remarkably ubiquitinated in the presence of p42 (Figure 4b). In contrast, silencing of p42 blocks p85 ubiquitination (Figure 4c). Thus, p42 facilitate degradation of p85 by the ubiquitin–proteasome pathway.

The p42 promotes p85 degradation through ubiquitin–proteasome system. (a) GFP-p42 (1 and 3 μg) was transfected into HEK293 cells, following exposure to 10 μM MG132 for 8 h. Immunoblotting was conducted to monitor p85, p42 or β-actin levels. (b) PC12 cells were co-transfected with HA-Ub and Flag-p85 and/or GFP-p42/p48 for 30 h and treated with MG132 (10 μM) for an additional 8 h. To obtain similar amount of immunoprecipitated Flag-p85, we transfected 0.5 μg of GFP-p42 while we transfected 1 μg of GFP or GFP-p48. Cell lysates were immunoprecipitated with anti-Flag and immunoblotted with anti-HA antibody. (c) HEK 293T cells were co-transfected with Flag-p85 and HA-Ub along with N-si-p48 or si-Ebp1 following exposure of MG132. Ubiquitinated p85 was detected by immunoblotting with anti-HA antibody. (d) Cells were transfected with various Flag-p85 fragments with or without GFP-p42, and p85 levels were determined by anti-Flag antibody (left). Densitometry analysis (right). **P<0.005
To further evaluate the specificity of p85 degradation by p42, we monitored the degradation of various truncations of p85. Interestingly, iSH2 and 3SH2 truncations that possess the iSH2 domain were readily diminished in the presence of p42 (Figure 4d). However, diSH2 and SH3BH, which do not possess iSH2, were relatively less affected by p42-mediated degradation. Hence, our data indicate that p42 strongly promotes a reduction of p85 protein level by targeting the iSH2 domain that has been described as having the most oncogenic mutations of p85 and is the location of many truncations or deletions.26, 27, 28, 29
P42 recruits a chaperone-E3 ligase complex HSP70/CHIP for the degradation of p85 subunit
Although Cbl-b was introduced as an E3 ubiquitin ligase for p85 in T cells, it does not exert proteasomal degradation of p85.25 The identity of the E3 ligase responsible for p85 degradation and its biological significance has not yet been reported. Hence, our finding that p42 mediates proteasomal degradation of p85 is striking. Because C-terminal fragments (280–394) of p42 showed a sufficient and prominent effect on p85 reduction (Figure 5a), we hypothesized that p42 recruits E3 ligase for p85 through its C-terminal domain (280–394) and screened C-terminal fragment (280–394) binding partners. Using mass-spectrometric analysis, we identified molecular chaperone HSP70 and co-chaperone E3 ligase CHIP that direct chaperone substrates for ubiquitination and proteasomal degradation as p42 interacting proteins (Figure 5b).

The p42 recruits a chaperone-E3 ligase complex HSP70/CHIP for p85 degradation. (a) HEK293T cells were transfected with various GFP-Ebp1 fragments, and p85 levels were examined by immunoblotting. (b) p42 interacts with HSP70 and CHIP. 293T cells were transfected with GST-Ebp1 (280–394 a.a.). After GST pull-down assay, the samples were subjected to silver staining according to the manufacturer’s instructions and applied for mass-spectrometry. (c) GST-pull down analysis was performed and immunoblotted with the indicated antibodies (left). 293T cell lysates were immunoprecipitated with anti-HSP70 antibody (upper) or anti-CHIP antibody (bottom) and immunoblotting with anti-Ebp1 or anti-N-Ebp1 antibody (right) (d) knockdown experiment with si-HSP70. (e and f) Ubiquitinated p85 was detected by immunoblotting with anti-HA antibody from Flag-p85- and HA-Ub-transfected PC12 cells. To obtain similar amount of immunoprecipitated Flag-p85, we transfected 0.5 μg of GFP-p42 while we transfected 1 μg of control. (g) p85 levels were monitored after co-transfection with different combinations of Myc-p42, GST-CHIP or HSP70. (h) Endogenous p85 levels were determined from 293T cells silenced with SCR, si-Ebp1 or si-CHIP (upper). Densitometry analysis (bottom). **P<0.005 versus control. (i) Endogenous protein levels were examined in the mouse embryonic fibroblast (MEF) CHIP (−/−) or CHIP (+/+) cells against anti-p85, anti-Ebp, anti-CHIP or anti-β-actin antibodies. (j) MEF CHIP (−/−) or CHIP (+/+) cells were co-transfected with Flag-p85 and HA-ub along with GFP-p42 and then exposed to MG132 for 8 h. (k) CHIP (−/−) cells was transfected with Myc-CHIP with or without p42 and immunoblotting was performed as indicated. (l) PI3K activity assay with GFP-p42- or Myc-CHIP-transfected cell lysates. *P<0.05 and **P<0.005
We confirmed that not only transfected but also endogenous p42 physically forms a complex with endogenous HSP70 and CHIP (Figure 5c), and this p42–HSP70 interaction is a prerequisite for the tricomplex formation, as it has been shown that depletion of HSP70 negates the association between p42 and CHIP (Figure 5d). This notion was supported by the finding that the deletion mutant of the TPR domain in CHIP or the K30A mutant, which is critical for association with HSP70, is unable to bind p42. However, the E3 ligase activity of CHIP is not necessary for interaction with p42, demonstrating that a deletion mutant of Ubox or the H260Q mutant form with no E3 ligase activity of CHIP is still able to associate with p42 (Supplementary Figures 4a and b), reflecting that p42 may not be an E3 ligase substrate of CHIP. Unexpectedly, we could not detect any direct interaction between p85 and HSP70 or p85 and CHIP (Supplementary Figures 4c and d). Conceivably, p42 couples p85 in close proximity to the catalytic site of the HSP70/CHIP complex, thereby inducing degradation of p85.
We next explored whether CHIP physiologically functions as an E3 ligase for p85 degradation. Silencing of CHIP completely abolished p85 ubiquitination (Figure 5e), whereas co-transfection of CHIP with p42 promoted p85 ubiquitination (Figure 5f), indicating that CHIP is a genuine ubiquitin ligase for p85 protein degradation. Interestingly, overexpression of CHIP or HSP70 alone does not affect p85 protein level without p42 expression, whereas co-transfection of CHIP/HSP70 with p42 notably decreased p85 protein level (Figure 5g). In contrast, silencing of CHIP rescued p85 protein, and silencing of both CHIP and p42 enhanced p85 protein level, although the synergistic effect of p42 depletion was limited (Figure 5h), reflecting the fact that endogenous p42 level is very low in most cell types2, 3 (Supplementary Figure 5a).
Endogenous p85 protein level was relatively higher in CHIP (−/−) mouse embryonic fibroblasts (MEFs) compared with that in CHIP (+/+) MEFs (Figure 5i). Accordingly, p85 ubiquitination was enhanced in CHIP(+/+) MEFs upon p42 expression but not in CHIP (−/−) MEFs (Figure 5j), confirming that CHIP is required for p42-mediated p85 degradation. Forced re-expression of CHIP or p42 alone in CHIP (−/−) MEFs failed to exert the diminution of p85; however, co-transfection of CHIP with p42 markedly decreased the endogenous p85 level (Figure 5k). Consequently, coexpression of p42/CHIP robustly alleviated PIP3 production activity of PI3K (Figure 5l and Supplementary Figure 5b). These data suggest that p42 inhibits PI3K activity, leading to ubiquitination and proteasomal degradation of p85 protein coupled with the HSP70/CHIP complex.
P42 represses tumorigenesis in vivo through downregulation of p85
Since our previous report showed that most of glioblastoma multiform (GBM) patients and GBM cell lines revealed undetectable protein expression of p42 and overexpression of p42 prohibited brain tumor growth24 (Supplementary Figures 6a and b), and our immunoblotting and immunohistochemical analysis with CHIP-deficient (CHIP-KO) mouse brain displayed high level of p85 protein compared with that of CHIP-WT mouse (Figures 6a and b and Supplementary Figure 6g) as well as the elevated PI3K activity shown by phospho-Akt levels (Figure 6a), we explored whether tumor-suppression function of p42 is involved in its new ability to link CHIP/HSP70 to p85 in vivo. We first generate mouse xenograft model with subcutaneous injection of U251 glioma cells into nude mice and monitored tumor development. After tumor development, we injected mice with adeno-associated virus (AAV) that expresses control GFP, p42-GFP or CHIP-GFP (Supplementary Figures 6c and d). Mice injected with p42-AAV or simultaneous expression of p42-AAV and CHIP-AAV after implantation of U251 tumor cells developed much smaller tumors than mice injected with the control vector (Figures 6c and d and Supplementary Figure S6e). Immunohistochemical analysis and immunoblotting of the tumor regions revealed low-intensity p85 expression in p42-AAV injection groups (Figure 6e and Supplementary Figure 6f and g), suggesting that p42 inhibits tumor growth through downregulation of p85, permitting access of HSP70/CHIP in vivo.

The p42 represses tumorigenesis in vivo through downregulation of p85. (a and b) Endogenous protein levels were examined in the mouse brain of CHIP-KO or CHIP-WT by immunoblotting and immunohistochemistry. The paraffin blocks were stained with anti-CHIP, anti-p85 antibody. Arrows indicate stained cells. Scale bar: 40 μm. (c and d) U251 cells (5 × 106) were injected subcutaneously into the nude mice. After tumor size reached 5 mm diameter, three groups of viral particles of AAV-GFP, AAV-p42 or AAV-p42 and AAV-CHIP were administrated to tumor region (as described in the Materials and Methods). After 21 days of AAV injection, animals were killed and their tumors were removed. Tumor volumes were measured from bi-dimensional measurements every 3 days and compared. The values shown are mean±S.E.M. **P<0.005 versus vector alone. Tumor weights were measured at 21 days after viral injection. Scale bar: 10 mm. (e) Representative tumor image of mice models were stained with hematoxylin and eosin (H&E, top). Proliferative cells were assessed by staining with an anti-PCNA antibody. Arrows indicate the PCNA-positive stained cells (middle). The expression level of p85 was stained with anti-p85 antibody. Arrows indicate p85-positive stained cells (bottom). Scale bar: 20 μm
Discussion
Despite expanding interest in the possible differential roles of two Ebp1 isoforms, p48 and p42, in tumorigenesis, the mechanisms by which p42 contributes to tumor suppression have not yet been fully characterized. We previously provided the molecular mechanism by which p48 functions as an oncogenic protein in glioma through negative regulation of p53.24 In an effort to determine how p42 functions as a tumor suppressor, in this study, we identified a unique degradation mechanism of the regulatory domain of PI3K, the p85 subunit, that is regulated by p42, permitting the access of HSP70/CHIP. Moreover, we verified that overexpression of p42 or co-expression of p42 with CHIP notably inhibited tumor growth and downregulated p85 protein expression in mouse xenograft model (Figures 6c–e and Supplementary Figure 6f).
It is striking that overexpression of p42 decreased p85 protein level because no degradation of p85 has yet been reported. One well-characterized E3 ligase that promotes ubiquitination of p85 is Casitas B-lineage lymphoma (Cbl) protein in immune cells, but Cbl does not exert p85 degradation.25, 30 Human Bre1, an E3 ligase for H2B monoubiquitination, binds p53 and enhances activator-dependent transcription.31 In previous studies, p42 was a target of hBre1 E3 ligase,3 as many of tumor suppressors are downregulated by UPS. However, p42 appears not to link hBre1 for the degradation of p85 (Supplementary Figure 7). Conversely, although p42 physically interacts with HSP70/CHIP, a decrease in p42 protein is very limited. Probably, it concedes the fact that HSP70/CHIP requires p42 for ablation of p85.
The reason why we could not detect the interaction between HSP70/CHIP and p85, may be that the interaction occurs very temporally to the iSH domain of p85 that displayed prominent disappearance (Figure 4d) when p42 supplied the HSP70/CHIP complex, exerting immediate degradation of iSH2. Presumably, as most of the tumor cells exhibit an extremely low level of p42 protein, p85 may not have had access to the HSP70/CHIP-mediated ubiquitination and degradation in tumors, allowing higher activation of PI3K. In agreement with this, p85 ubiquitination and degradation occurred only when p42 was concomitantly expressed, and p42-mediated reduction of p85 protein was augmented by coexpression of CHIP and was attenuated by depletion of CHIP (Figures 5g and h). These findings may account for recent studies on cancer-derived mutations of p85. For example, the cSH2 deletion mutant of p85, which disrupts p42 binding, was found in a Hodgkin’s lymphoma-derived cell line (CO), and PI3K is constitutively active in these cells.32 In addition, frequent oncogenic mutations in p85 in colon cancer occur in the iSH domain, a major target of p42-mediated degradation. Presumably, p42 serves in normal cells as a sensor of oncogenic stress that represses aberrant activation of PI3K by providing UPS to the iSH2 domain of p85 through direct interaction with the cSH2 domain of p85 but permits oncogenic activation of PI3K by disrupting the binding of UPS because of faint expression of p42 in tumor cells (Supplementary Figure S4a). It remains to be determined whether any of these oncogenic mutations in p85 prevent p85 degradation by p42-mediated UPS. In addition, 3SH2 truncation that includes nSH2/iSH2/cSH2 domain appeared to slightly enhance the binding affinity of p85 to p42 compared with iSH2/cSH2-containing fragment (Figure 1d), suggesting the possibility that nSH2/iSH2/cSH2 fragment is more reluctant for p42-mediated degradation than iSH2/cSH2 fragment because of an addition of nSH2 domain or nSH2 domain might mask iSH2 domain to delay its rapid degradation in the presence of p42 expression as it has been defined that nSH2 domain rotates much more freely within nSH2/iSH2 fragment. It will be worth to study further that the identification of regulatory role of nSH2 domain if it can provide the precise regulatory mechanism of p85 subunit regulation by tumor suppressor.
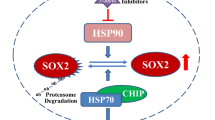
Our discovery that tumor-suppressor p42 partners with HSP70/CHIP, forming novel E3 ligase complex for p85 to elicit p85 degradation (Figure 7), has important ramifications for our understanding of the cell intrinsic control of tumor suppression using one alternative spliced isoform whereas the other is employed for tumor growth. It can also account for recent studies on cancer-derived mutations of p85 and provides a new concept of PI3K regulation in normal and cancer cells. It will be important to study the repression of p42 protein expression in cancer cells, and as p42 shows the inhibitory role of PI3K pathway through its C-terminal domain, small molecule that mimics the C-terminal domain of p42 may have therapeutic potential.

Schematic diagram of the cell signal pathway that p42 utilizes to suppress tumor growth by controlling PI3K activity through UPS. As most of the tumor cells exhibit an extremely low level of p42 protein, p85 may not have had access to the HSP70/CHIP-mediated ubiquitination and degradation in tumors, allowing higher activation of PI3K. However, once p42 protein level is upregulated, p42 provides p85 to HSP70/CHIP complex for the degradation of p85 by interacting with p85 and HSP70/CHIP, thereby inhibiting PI3K activity
Materials and Methods
Cell cultures
HEK293T, MDA-MB-231, U251 and MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 100 U of penicillin/streptomycin. Primary MEF CHIP (+/+) and CHIP (−/−) cells were a kind gift of Dr. Chin Ha Chung (Seoul National University, Seoul, Korea) and were maintained in high glucose DMEM supplemented with 10% FBS and antibiotics. T47D and MDA-MB-231 cells were a gift of Dr. Chang-Woo Lee (Sungkyunkwan University, Suwon, Korea). T47D cells were cultured in RPMI-1640 supplemented with 10% FBS, 10 μg/ml insulin and 100 U of penicillin/streptomycin. PC12 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% FBS, 5% horse serum and 100 U of penicillin/streptomycin at 37°C under a 5% CO2 atmosphere.
Cell culture and establishment of Ebp1 stable expression cell lines
The human malignant glioma cell line, U251 (purchased from the American Type Culture Collection, Manassas, VA, USA), was maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated FBS and incubated in a humidified incubator at 37°C with 5% CO2. Plasmid transfection was performed using electroporator (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For selection of stable clone, pEGFP-p48, pEGFP-p42 and only pEGFP vectors were transfected into several glioma cells and incubated for 48 h at 37°C. These cells were changed in medium containing G418 (600 μg/ml). After 2 or 3 weeks, G418-resistant colonies were isolated using FACS sorter or colony picking method, and individual colonies were further cultured.
Antibodies, siRNA and chemicals
Anti-p85 and anti-CHIP antibodies were acquired from Cell Signaling (Boston, MA, USA). Anti-GFP, anti-GST, anti-MYC, anti-Actin and anti-p110 antibodies were acquired from Santa Cruz Biotechnology Technology, Inc. (Dallas, TX, USA). Anti-FLAG antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA). Alexa Fluor 594 goat anti-mouse and Alexa Fluor 350 donkey anti-mouse secondary antibodies were obtained from Molecular Probes (Eugene, OR, USA). Anti-HSP70 and Ebp1 antibodies were obtained from Abcam (Cambridge, MA, USA). The siRNAs for si-Ebp1 (5′-UGUAAAUAGUGGUUCUCUGUCCUGCAU-3′), si-CHIP (5′-ACCACGAGGGUGAUGAGGUAA-3′) and si-p85 (5′-CUGAGUAUCGAGAAAUUGAUU-3′) were obtained from Genolution (Seoul, Republic of Korea). The siRNA for Si-HSP70 (5′-GAAGGACGAGUUUGAGCACAA-3′) was obtained from Cosmogentech (Seoul, Republic of Korea). The siRNA for N-si-p48 (duplex oligonucleotide, 5′-CAGCGAUAGUUUGCUCCUGUUGCUCGU-3′ and 3′-GTCGCUAUCAAACGAGGACAACGAG-5′) was obtained from Integrated DNA Technologies, Inc. (Coralville, IA, USA). A PI3K ELISA assay kit was purchased from Echelon Biosciences (San Jose, CA, USA), and a silver staining kit was purchased from GE Healthcare (Uppsala, Sweden). EGF was purchased from R&D Systems (Minneapolis, MN, USA). CHX was purchased from Duchefa Biochemie (Haarlem, The Netherlands). MG132 was obtained from Sigma (St. Louis, MO, USA).
Constructions of recombinant DNA and AAV
The various fragments of p85 and p110 subunits of PI3K were cloned into pcDNA3-Flag. HSP70 and E3 ligase CHIP were cloned into pcDNA-GST. Myc-tagged CHIP fragments were a gift from Dr. Chin Ha Chung (Seoul National University). HA-tagged hBre1 was a gift from Dr. Keqiang Ye (Emory University, Atlanta, GA, USA). Ebp1 p42 and CHIP were inserted in the AAV2-IRES-GFP vector and packed in the 293 AAV cell line for high titer production of AAV Cell Biolab (San Diego, CA, USA). AAV packaging service was provided from KIST (Korea Institute of Science and Technology, Seoul, Republic of Korea).
Co-immunoprecipitation assay and in vitro binding assay
Co-immunoprecipitation assays were performed as previously described.33 Cells were rinsed with phosphate-buffered saline (PBS) and lysed in buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 10 mM β-glycerolphosphate, 1 mM phenylmethlysulfonyl fluoride (PMSF) and protease cocktail (Calbiochem, San Diego, CA, USA)). Cell lysates (0.5 to 1 mg of protein) were mixed with primary antibody with protein A/G beads and incubated for 3 h at 4°C with gentle agitation. The beads were then washed in lysis buffer, mixed with 2 × SDS sample buffer, boiled and analyzed by immunoblotting.
GST pull-down assay
Cells were rinsed with PBS and lysed in buffer as described above. Cell lysates (0.5 to 1 mg of protein) were mixed with glutathione-sepharose beads and incubated for 3 h at 4°C with gentle agitation. The beads were then washed in lysis buffer, mixed with 2 × SDS sample buffer, boiled and analyzed by immunoblotting.
Immunofluorescence
Cells grown on coverslips in 24-well plates were fixed in 4% paraformaldehyde for 15 min, permeabilized in PBS containing 0.25% Triton X-100 for 10 min and blocked in 2% BSA for 30 min. Cells were immunostained using primary antibodies and the appropriate Alexa Fluor 594 goat anti-mouse and Alexa Fluor 350 donkey anti-mouse secondary antibodies. Nuclei were counterstained with Hoechst. Immunostained images were acquired using a laser scanning confocal microscope LSM 710 META Duoscan (Carl Zeiss, Oberkochen, Germany) equipped with a 40 × water immersion C-Apochromat objective (Carl Zeiss) at room temperature (22°C). The confocal microscope was controlled using LSM software version 4.2 (Carl Zeiss).
Proliferation, invasion and colony formation assay
Cells were plated (5 × 104 cells per 60-mm dish) and viable cell numbers were counted as indicated time using BrdU cell proliferation kit according to the manufacturer’s manual (Cell Signaling Technology, Inc.). Invasion assays were performed as previously described34 using Matrigel invasion assay Kit (BD Bioscience, Inc., Franklin Lakes, NJ, USA). For colony-forming assay, cells were seeded on 35-mm dishes in 0.35% bacto-agar (supplemented with complete medium; Becton, Dickinson and Co., Franklin Lakes, NJ, USA) and cultured for 4 weeks. The cells were fixed and stained with crystal violet, and then counted in random five fields. Each value from these indicated assays represents the mean±S.E.M. of triplicate measurements from three independent experiments.
ELISA for PI3K activity
Cells were transfected with GFP control (7 μg), GFP-p48 (7 μg) or GFP-p42 (12 μg). After 24 h of transfection, cell extracts (1 mg) were subjected to immunoprecipitation with the anti-p110 antibody. PI3K activity was evaluated with a PI3K competitive ELISA kit (Echelon Biosciences) according to the manufacturer’s protocol. Briefly, 60 μl of kinase reaction samples were added to each well, and 60 μl/well of the above diluted PIP(3,4,5)P3 detector was also added. Plates were incubated at room temperature for 60 min. After incubation, we transferred 100 μl of the reaction samples from the incubation plate to the detection plate. We sealed the plates and incubated them for 60 min at room temperature. Plates were then washed three times with washing buffer and incubated with 100 μl of the secondary detector at room temperature for 30 min. Plates were then washed three times with washing buffer and incubated with 100 μl of the TMB substrate solution at room temperature for 10–20 min. The ELISA was stopped with 50 μl/well stop solution and read at OD 450 nm. Each value from these indicated assays represents the mean±S.E.M. of triplicate measurements from three independent experiments.
TLC analysis for PI3K activity
Cells were immunoprecipitated by the anti-p110 antibody and washed with the following buffers: three times with Buffer A (PBS, 1% NP-40 and 1 mM DTT), two times with Buffer B (PBS, 0.5 M LiCl and 1 mM DTT) and two times with Buffer C (10 mM Tris-HCl, pH 7.4, 0.1 M NaCl and 1 mM DTT). After 5 min at room temperature, the immunoprecipitates were incubated with the reaction buffer (20 mM HEPES (pH 7.5), 20 mM MgCl2, 100 μM ATP containing, 10 μCi/ATP(32P)) for an additional 10 min at 37°C. The reaction was stopped by adding 20 μl of 6N HCl and 130 μl of chloroform/methanol (1 : 1). After vortexing, 30 μl of the lower chloroform phase was separated by TLC, and the radiolabeled spots were visualized by autoradiography.
Stub1 knockout mice
We have received Stub1 knockout mice from Riken animal center (Tsukuba, Japan).35 Stub1 knockout mice were maintained on genetic background of C57BL/6.
Immunohistochemistry
The IHC staining was conducted as described in previous report.36 Briefly, paraffin-embedded tissues were cut into 4 μm thick sections. The sections were then deparaffinized in xylene 3 times for 10 min each and rehydrated in a graded ethanol series (100, 95, 75 and 50%) before incubation for 10 min with 3% hydrogen peroxide solution. The sections were washed in PBS. The optimal dilution was determined for each antibody by serial dilutions. A negative control was set up for each antibody by omitting the primary antibody (Supplementary Figure 6g) and specificity of primary antibodies was verified by immunoblotting throughout this research and also well confirmed by other groups as well.37, 38, 39, 40, 41, 42, 43, 44, 45 The primary antibody incubation was completed at 4°C overnight in a humidified chamber with the following antibodies: mouse monoclonal antibody PI3K-p85 (sc-1673, Santa Cruz Biotechnology; dilution 1 : 150),37, 38, 39, 40 mouse monoclonal antibody CHIP (sc-133066, Santa Cruz Biotechnology; dilution 1 : 200)41, 42, 43 and mouse monoclonal antibody PCNA (Calbiochem; dilution 1 : 500).44, 45 After incubation, each section was washed with PBS and incubated with the secondary biotinylated goat-anti mouse antibody (Vector Laboratories, Burlingame, CA, USA) for 30 min at room temperature. After being washed in PBS, the sections were incubated with streptavidin conjugated with horseradish peroxidase (VECTASTAIN ABC kit, Vector Laboratories). Tissue sections were washed 3 times for 5 min each in PBS buffer and covered with 3,3′-diaminobenzidine solution (DAB SUBSTRATE KIT, Vector Laboratories) for 5 min. Finally, the specimens were counterstained with hematoxylin. Negative control slides were incubated with the antibody diluents instead of the primary antibody.
Mouse xenograft model
All procedures were approved by the Sungkyunkwan University School of Medicine Institutional Animal Care and Use Committee. Briefly, 6- to 8-week-old athymic nude (nu/nu) mice were housed in laminar-flow cabinets under specific pathogen-free conditions. For the establishment of glioma model, animals were anesthetized by intraperitoneal (i.p.) injection with 12 mg/kg of xylazine (Rompun; Cutter Laboratories, Shawnee, KS, USA) and 30 mg/kg of ketamine (Yuhan Ketamine; Yuhan Co., Ltd, Seoul, Korea) and were held in a stereotactic frame with an ear bar. Human glioblastoma cell lines (U251) cells stably transfected with GFP only vector, GFP-p48 Ebp1 or GFP-p42 Ebp1 (1 × 105 cells in a volume of 3 μl PBS, n=5 mice per group) were injected slowly into the brain (2 mm lateral and 1 mm anterior to bregma, at a 3 mm depth from skull surface) with a Hamilton syringe. At 14 days after stable glioma cell inoculation, the animals were killed and fixed by intracardiac perfusion of 4% paraformaldehyde in PBS, and the brains were removed. The presence of tumor in the brain was evaluated by serial coronal cutting with 1 mm thickness from the tumor cell inoculation site. The midcoronal sections of the whole tumors were processed for histological and immunohistochemical analyses. For the construction of viral-infected tumor model, U251 cells (5 × 106 cells) were injected subcutaneously into the right flank of each nude mouse (total 15 mice). The animals were randomized into three groups (n=5 per each group) when the solid tumor size reached 5 mm diameter. Then, 0.5 μl of viral particles expressing AAV-GFP-control, AAV-GFP-p42 or AAV-GFP-CHIP with AAV-GFP-p42 (1011 GC/ml) were administrated via intraperitoneal injection. At 3 weeks after viral infection, a western blot of the tissue at the injected site (2 mm × 2 mm × 2 mm) was performed (Supplementary Figure S5f). The tumor size was monitored every 2 days using calipers, and tumor volume (V) was calculated using the formula V= (ab)/2, in which a is the longest and b is the shortest diameter of the tumor (Figure 6c). Coronal sections of the whole tumors were processed for histological and immunohistochemical analyses. Representative tumor of AAV-GFP-control, AAV-GFP-p42 or AAV-GFP-CHIP/p42 Ebp1 animals was stained with H&E (Figure 6e, top). Proliferative cells were assessed by staining with an anti-PCNA antibody. The p85 expression was determined by staining with an anti-p85 antibody. Arrows indicate the PCNA-positive cells (middle) and p85-positive cells (bottom). Scale bar=10 μm.
Statistical analysis
Data are expressed as mean±S.E.M. of triplicate measurements from three independent experiments. Statistical analysis was performed by Sigmaplot Statistical Analysis Software (Systat software, San Jose, CA, USA). All studies were performed in a blinded manner. Statistical significance was defined as *P<0.05 and **P<0.005.
Abbreviations
- Ebp1:
-
ErbB3-binding protein 1
- PI3K:
-
phosphathidyl inositol 3-kinase
- SH:
-
Src-homology
- HSP70:
-
heat shock protein 70
- CHIP:
-
carboxy terminus of heat shock protein 70 (Hsp70)-interacting protein
- UPS:
-
ubiquitin–proteasome system
- MEF:
-
mouse embryonic fibroblast
- AAV:
-
adeno-associated virus
References
Ahn JY, Liu X, Liu Z, Pereira L, Cheng D, Peng J et al. Nuclear Akt associates with PKC-phosphorylated Ebp1, preventing DNA fragmentation by inhibition of caspase-activated DNase. EMBO J 2006; 25: 2083–2095.
Liu Z, Ahn JY, Liu X, Ye K . Ebp1 isoforms distinctively regulate cell survival and differentiation. Proc Natl Acad Sci USA 2006; 103: 10917–10922.
Liu Z, Oh SM, Okada M, Liu X, Cheng D, Peng J et al. Human BRE1 is an E3 ubiquitin ligase for Ebp1 tumor suppressor. Mol Biol Cell 2009; 20: 757–768.
Monie TP, Perrin AJ, Birtley JR, Sweeney TR, Karakasiliotis I, Chaudhry Y et al. Structural insights into the transcriptional and translational roles of Ebp1. EMBO J 2007; 26: 3936–3944.
Zhang Y, Hamburger AW . Heregulin regulates the ability of the ErbB3-binding protein Ebp1 to bind E2F promoter elements and repress E2F-mediated transcription. J Biol Chem 2004; 279: 26126–26133.
Zhang Y, Akinmade D, Hamburger AW . Inhibition of heregulin mediated MCF-7 breast cancer cell growth by the ErbB3 binding protein EBP1. Cancer Lett 2008; 265: 298–306.
Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC . Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci 2005; 30: 194–204.
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007; 318: 1744–1748.
Elis W, Lessmann E, Oelgeschlager M, Huber M . Mutations in the inter-SH2 domain of the regulatory subunit of phosphoinositide 3-kinase: effects on catalytic subunit binding and holoenzyme function. Biol Chem 2006; 387: 1567–1573.
Amzel LM, Huang CH, Mandelker D, Lengauer C, Gabelli SB, Vogelstein B . Structural comparisons of class I phosphoinositide 3-kinases. Nat Rev Cancer 2008; 8: 665–669.
Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G et al. PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J 1994; 13: 511–521.
Klippel A, Escobedo JA, Hirano M, Williams LT . The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol Cell Biol 1994; 14: 2675–2685.
Wolf DH, Hilt W . The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta 2004; 1695: 19–31.
Hershko A, Ciechanover A . The ubiquitin system. Annu Rev Biochem 1998; 67: 425–479.
Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP et al. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013; 32: 3101–3110.
Cyr DM, Hohfeld J, Patterson C . Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci 2002; 27: 368–375.
Murata S, Chiba T, Tanaka K . CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int J Biochem Cell Biol 2003; 35: 572–578.
Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J et al. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem 2001; 276: 42938–42944.
Murata S, Minami Y, Minami M, Chiba T, Tanaka K . CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2001; 2: 1133–1138.
Gu J, Tamura M, Yamada KM . Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol 1998; 143: 1375–1383.
Kim CK, Lee SB, Nguyen TL, Lee KH, Um SH, Kim J et al. Long isoform of ErbB3 binding protein, p48, mediates protein kinase B/Akt-dependent HDM2 stabilization and nuclear localization. Exp Cell Res 2012; 318: 136–143.
Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993; 72: 767–778.
Holt KH, Olson L, Moye-Rowley WS, Pessin JE . Phosphatidylinositol 3-kinase activation is mediated by high-affinity interactions between distinct domains within the p110 and p85 subunits. Mol Cell Biol 1994; 14: 42–49.
Kim CK, Nguyen TL, Joo KM, Nam DH, Park J, Lee KH et al. Negative regulation of p53 by the long isoform of ErbB3 binding protein Ebp1 in brain tumors. Cancer Res 2010; 70: 9730–9741.
Fang D, Liu YC . Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol 2001; 2: 870–875.
Jimenez C, Jones DR, Rodriguez-Viciana P, Gonzalez-Garcia A, Leonardo E, Wennstrom S et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J 1998; 17: 743–753.
Jucker M, Sudel K, Horn S, Sickel M, Wegner W, Fiedler W et al. Expression of a mutated form of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkin's lymphoma-derived cell line (CO). Leukemia 2002; 16: 894–901.
Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res 2001; 61: 7426–7429.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–1812.
Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC . Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem 2001; 276: 4872–4878.
Hofmann BT, Jucker M . Activation of PI3K/Akt signaling by n-terminal SH2 domain mutants of the p85alpha regulatory subunit of PI3K is enhanced by deletion of its c-terminal SH2 domain. Cell Signal 2012; 24: 1950–1954.
Kim J, Hake SB, Roeder RG . The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell 2005; 20: 759–770.
Deng Y, Li Y, Fan X, Yuan W, Xie H, Mo X et al. Synergistic efficacy of LBH and alphaB-crystallin through inhibiting transcriptional activities of p53 and p21. BMB Rep 2010; 43: 432–437.
Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye K . PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem 2004; 279: 16441–16451.
Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R et al. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem 2005; 94: 1254–1263.
Kim EA, Hahn HG, Kim TU, Choi SY, Cho SW . Attenuation of beta-amyloid-induced neuroinflammation by KHG21834 in vivo. BMB Rep 2010; 43: 413–418.
Abou Youssif T, Fahmy MA, Koumakpayi IH, Ayala F, Al Marzooqi S, Chen G et al. The mammalian target of rapamycin pathway is widely activated without PTEN deletion in renal cell carcinoma metastases. Cancer 2011; 117: 290–300.
Gurney SM, Forster P, Just U, Schwanbeck R . Suppression of the PI3K subunit p85alpha delays embryoid body development and inhibits cell adhesion. J Cell Biochem 2011; 112: 3573–3581.
Korkolopoulou P, Levidou G, Trigka EA, Prekete N, Karlou M, Thymara I et al. A comprehensive immunohistochemical and molecular approach to the PI3K/AKT/mTOR (phosphoinositide 3-kinase/v-akt murine thymoma viral oncogene/mammalian target of rapamycin) pathway in bladder urothelial carcinoma. BJU Int 2012; 110 (11 Pt C): E1237–E1248.
Zhu Q, Youn H, Tang J, Tawfik O, Dennis K, Terranova PF et al. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene 2008; 27: 4569–4579.
Gao B, Wang Y, Xu W, Li S, Li Q, Xiong S . Inhibition of histone deacetylase activity suppresses IFN-gamma induction of tripartite motif 22 via CHIP-mediated proteasomal degradation of IRF-1. J Immunol 2013; 191: 464–471.
Sarkar S, Brautigan DL, Parsons SJ, Larner JM . Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene 2012; 33: 26–33.
Wen J, Luo KJ, Hu Y, Yang H, Fu JH . Metastatic lymph node CHIP expression is a potential prognostic marker for resected esophageal squamous cell carcinoma patients. Ann Surg Oncol 2013; 20: 1668–1675.
Bianchi MS, Hernando-Insua A, Chasseing NA, Rodriguez JM, Elias F, Lago N et al. Oligodeoxynucleotide IMT504 induces a marked recovery in a streptozotocin-induced model of diabetes in rats: correlation with an early increase in the expression of nestin and neurogenin 3 progenitor cell markers. Diabetologia 2010; 53: 1184–1189.
Suzuka I, Daidoji H, Matsuoka M, Kadowaki K, Takasaki Y, Nakane PK et al. Gene for proliferating-cell nuclear antigen (DNA polymerase delta auxiliary protein) is present in both mammalian and higher plant genomes. Proc Natl Acad Sci USA 1989; 86: 3189–3193.
Acknowledgements
We thank Dr. Chin Ha Chung, Seoul National University, Republic of Korea, for providing CHIP (−/−) MEF cells and DNA constructs. This research was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (1120090).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Author Contributions
HRK, K-HL, S-WC and JYA designed research; HRK, CKK, SBL, JS, KKK, KWP and J-YA performed research; HRK, K-HL, S-WC and J-YA analyzed data; J-YA wrote the paper.
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ko, H., Kim, C., Lee, S. et al. P42 Ebp1 regulates the proteasomal degradation of the p85 regulatory subunit of PI3K by recruiting a chaperone-E3 ligase complex HSP70/CHIP. Cell Death Dis 5, e1131 (2014). https://doi.org/10.1038/cddis.2014.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.79
Keywords
This article is cited by
-
CHIP promotes CAD ubiquitination and degradation to suppress the proliferation and colony formation of glioblastoma cells
Cellular Oncology (2023)
-
Cerebellar dysfunction and schizophrenia-like behavior in Ebp1-deficient mice
Molecular Psychiatry (2022)
-
The Role of the Ubiquitin Proteasome System in Glioma: Analysis Emphasizing the Main Molecular Players and Therapeutic Strategies Identified in Glioblastoma Multiforme
Molecular Neurobiology (2021)
-
The role of ubiquitination and deubiquitination in cancer metabolism
Molecular Cancer (2020)
-
Modeling the Bistable Dynamics of the Innate Immune System
Bulletin of Mathematical Biology (2019)
