
Abstract
The reduced clearance of amyloid-β peptide (Aβ) from the brain partly accounts for the neurotoxic accumulation of Aβ in Alzheimer’s disease (AD). Recently, it has been suggested that P-glycoprotein (P-gp), which is an efflux transporter expressed on the luminal membrane of the brain capillary endothelium, is capable of transporting Aβ out of the brain. Although evidence has shown that restoring P-gp reduces brain Aβ in a mouse model of AD, the molecular mechanisms underlying the decrease in P-gp expression in AD is largely unknown. We found that Aβ1–42 reduced P-gp expression in the murine brain endothelial cell line bEnd.3, which was consistent with our in vivo data that P-gp expression was significantly reduced, especially near amyloid plaques in the brains of five familial AD mutations (5XFAD) mice that are used as an animal model for AD. A neutralizing antibody against the receptor for advanced glycation end products (RAGE) and an inhibitor of nuclear factor-kappa B (NF-κB) signaling prevented the decrease in Aβ1–42-induced P-gp expression, suggesting that Aβ reduced P-gp expression through NF-κB signaling by interacting with RAGE. In addition, we observed that the P-gp reduction by Aβ was rescued in bEnd.3 cells receiving inductive signals or factors from astrocytes making contacts with endothelial cells (ECs). These results support that alterations of astrocyte–EC contacts were closely associated with P-gp expression. This suggestion was further supported by the observation of a loss of astrocyte polarity in the brains of 5XFAD mice. Taken together, we found that P-gp downregulation by Aβ was mediated through RAGE–NF-κB signaling pathway in ECs and that the contact between astrocytes and ECs was an important factor in the regulation of P-gp expression.
Similar content being viewed by others


Main
Alzheimer’s disease (AD) is a neurodegenerative disorder that is characterized by a progressive loss of cognitive function leading to dementia. The major pathological hallmark of AD is the deposition of neurotoxic amyloid-β peptide (Aβ) within the brain.1 The amyloid hypothesis proposes that the accumulation of Aβ is caused by an imbalance between Aβ production and clearance.2 Although genetic alterations increase the production of Aβ in rare familial AD, reduced Aβ clearance from the brain likely accounts for sporadic AD, which is much more common.3 The mechanisms that are involved in clearing Aβ from the brain include enzymatic degradation, perivascular drainage, and the most significant, active transport across the blood–brain barrier (BBB).4
The BBB regulates molecular exchanges at the interface between the blood and the brain.5 It plays a critical role in maintaining the brain microenvironment.6 The BBB, which is formed by cerebral endothelial cells (ECs) and which, interacts with astrocytes, neurons, pericytes, and the extracellular matrix, is organized into a neurovascular unit.7, 8 Although the relationship between BBB breakdown and AD pathology is unclear,9 it has been proposed that the BBB loses its Aβ clearing capability, thus increasing amyloid deposition in the outer capillary membrane and resulting in the distortion of the neurovascular unit with neuronal loss.10
Recently, it has been suggested that P-glycoprotein (P-gp), which is an ATP-driven efflux transporter that is highly expressed in the luminal membrane of the brain capillary endothelium, is also involved in the clearance of Aβ from the brain.11 P-gp, which is able to transport various kinds of substrates, has been shown to play an important role in clearing toxic substances in the brain and protecting it from harmful molecules in the circulation.12 Along with other BBB properties, P-gp expression is induced when ECs are in contact with astrocytes in vitro and in vivo.13, 14 ECs respond to inductive signals or factors from astrocytes that encircle the capillary endothelium.13
Several lines of evidence have shown that P-gp plays an important role in Aβ clearance. It has been shown in vitro that P-gp mediates the transport of Aβ and that blocking P-gp function reduces the clearance of Aβ.15, 16 In addition, cerebral Aβ deposition in elderly non-demented individuals has been demonstrated to be inversely correlated with brain capillary P-gp expression.17 Furthermore, in P-gp knockout mice, Aβ deposition is increased by the reduced efflux of Aβ,18 while it has been shown that restoring P-gp at the BBB reduces brain Aβ in a mouse model of AD.19 However, the molecular mechanisms underlying the decrease in P-gp expression that is observed in AD have not been identified. We found that Aβ decreased P-gp expression by increasing nuclear factor-kappa B (NF-κB) through an interaction with the receptor for advanced glycation end products (RAGE). Moreover, we observed that the P-gp reduction by Aβ was rescued by inductive signals or factors from astrocytes that made contact with ECs in bEnd.3 cells. These results suggested that alterations in astrocyte–EC contact in AD likely decrease P-gp expression by Aβ. Together, we identified a mechanism by which the Aβ–RAGE interaction mediated the downregulation of P-gp in the BBB by increasing NF-κB signaling in AD and that astrocyte–EC contact played a critical role in maintaining P-gp expression.
Results
P-gp expression was decreased in the brain capillaries of 5XFAD mice
A previous study has shown that 5XFAD mice exhibit massive amyloid deposition from 2 months of age and memory deficits from 4 to 6 months of age.20, 21 Because P-gp plays a role in the efflux of Aβ from the brain to the periphery, we used the brain slices of 6-month-old 5XFAD mice and isolated the brain capillaries in order to examine the levels of expression of P-gp within the cerebral capillaries of 5XFAD mice. P-gp expression was significantly reduced in the isolated brain capillaries of 5XFAD mice compared to wild-type mice (Figures 1a and b). Because the expression of GLUT-1 is limited to the endothelium,22 we checked the GLUT-1 levels and confirmed the decreased levels of GLUT-1 in 5XFAD mice compared to wild-type mice in order to evaluate whether the brain capillaries were efficiently isolated in the brains of 5XFAD mice. In addition, in order to verify each individual wild-type and 5XFAD mouse, the isolated capillaries were prepared for immunoblot analysis with an anti-Aβ antibody (6E10), and only Aβ levels were detected in the brains of 5XFAD mice (Figure 1a). In order to identify the exact expression pattern of P-gp, we costained for P-gp and amyloid plaques with an anti-P-gp antibody and the anti-Aβ antibody, 4G8. P-gp was hardly detected near the amyloid plaque deposits in 5XFAD mice (Figure 1c, the parts shown in the white dotted circles), while it was clearly detected along the brain capillaries far from the amyloid plaque deposits in 5XFAD mice (Figure 1c, the parts shown in the yellow dotted circles) and wild-type mice (Figure 1c, yellow arrow heads). In order to clearly recognize the change in P-gp expression near the amyloid plaques, we used super-resolution SIM. The reconstructed three-dimensional (3D) SIM images showed that P-gp was expressed along the endothelium in the brains of wild-type mice (Figure 1d, yellow arrow heads). Also, P-gp expression was markedly disconnected near the amyloid plaque deposits in 5XFAD mice (Figure 1d, white arrow heads) compared to wild-type mice. These results implied that P-gp expression was decreased near the amyloid plaques in the cerebral capillaries of 5XFAD mice.

P-glycoprotein (P-gp) expression was decreased in the brain capillaries of five familial Alzheimer’s disease mutations (5XFAD) mice. (a and b) P-gp protein levels were measured by performing western blotting on total cerebral homogenates and cerebral capillaries that were isolated from the brains of wild-type and 5XFAD mice. β-Actin was used as a loading control. (c) Confocal microscopic analysis of P-gp and amyloid plaques in cortical sections (30 μm) from 6-month-old control and 5XFAD mice (n=3 for each). The amyloid plaques were labeled with the specific anti-amyloid-β peptide (Aβ) antibody (green; 4G8). P-gp (red) was clearly detected along the capillaries of the wild-type mice and far from the amyloid plaque deposits in 5XFAD mice (yellow arrow heads and the parts shown in the yellow dotted circles), but hardly detected near the amyloid plaque deposits in 5XFAD mice (the parts shown in the white dotted circles). Scale bars=10 and 30 μm. (d) Three-dimensional-structured illumination microscopy (3D-SIM) image of P-gp and amyloid plaques in the brains of wild-type and 5XFAD mice (n=3 for each). Amyloid plaques and P-gp were identified by the immunofluorescent detection of the 4G8 (green) and anti-P-gp antibody (red). Each panel shows different regions in the brains. The arrow heads indicate P-gp expression. Scale bar=2 μm
Aβ1–42 decreased P-gp expression in bEnd.3 cells
The ECs lining the cerebral microvessels contain P-gp and various ATP binding cassette efflux transporters.23 The levels of P-gp in the brain vasculature appear to be lower in the hippocampus of AD brains than in those of non-demented subjects.24 In order to determine the effectors underlying the decreased P-gp levels of expression near the amyloid plaque deposits that were seen in the capillaries of 5XFAD mice in the above results, we examined the changes in P-gp expression by Aβ1–42 in a monolayer culture of bEnd.3 cells.25 A western blot analysis showed that P-gp protein levels were significantly decreased after 24 h of 5 μM of Aβ1–42 treatment in bEnd.3 cells (Figures 2a and b). In addition, by performing an immunofluorescence analysis after Aβ1–42 treatment, P-gp immunoreactivity showed a weak staining pattern (Figure 2c), and the red peak value indicating the fluorescence intensity of P-gp fell in the bEnd.3 cells after 24 h of Aβ1–42 treatment compared to the control cells in the representative fluorescence intensity profiles (Figure 2d). The peak fluorescence intensity was significantly reduced in Aβ1–42-treated bEnd.3 cells (Figure 2e). The peak fluorescence intensity values from 10 cells per group of three independent preparations were analyzed. These results suggested that Aβ1–42 decreased the protein levels of P-gp in bEnd.3 cells.

Aβ1–42 decreased P-gp expression in bEnd.3 cells. (a) P-gp expression levels were measured by a western blot analysis in bEnd.3 cells that were incubated with various doses of Aβ1–42 for 24 h. Tubulin was used as a loading control. (b) A histogram represents the densitometric analysis of blots, with the data expressed as mean±standard error of the mean (S.E.M.; n=4; **P<0.01 versus Aβ 0 μM). (c) Confocal microscopic analysis of the P-gp intensity in bEnd.3 cells that were treated with 5 μM of Aβ1–42 for 24 h. The nuclei were labeled with 4′-6-diamidino-2phenylindole (DAPI; blue), and the P-gp immunoreactivity was labeled in red. Scale bar=20 μm. (d and e) Fluorescence intensity profile of each imaged cells that was determined by drawing lines parallel to the axis of the cell. The x-axis shows the length of the line, which was ∼30 μm in total, and the y-axis shows the intensity. The red line shows the intensity of P-gp, and the blue line shows that of DAPI
Aβ1–42 decreased P-gp expression through NF-κB activation
It has been reported that the expression of the p50 subunit of NF-κB is increased in the cerebral vessels and neurons of subjects with AD.26, 27 In order to examine whether NF-κB signaling mediated the Aβ1–42-induced decrease in P-gp expression in bEnd.3 cells, we cotreated bEnd.3 cells with Aβ1–42 and the inhibitor BAY 11-7082, which inhibits the NF-κB pathway by suppressing the phosphorylation of IκB.28 In the presence of 5 μM of BAY 11-7082, the Aβ1–42-induced decrease in P-gp expression was significantly attenuated (Figures 3a and b). Further immunofluorescence analysis by confocal microscopy also showed attenuation by 5 μM of BAY 11-7082 on the Aβ1–42-induced decrease in P-gp expression (Figure 3c). After treatment with Aβ1–42 in the presence of BAY 11-7082, peak fluorescence intensity was preserved than Aβ1–42-treated bEnd.3 cells (Figures 3d and e). In order to assess whether BAY 11-7082 inhibited IκB phosphorylation efficiently, the degree of phosphorylation on IκB was examined by western blotting. There was a significant reduction in the phosphorylated form of IκB (p-IκB) in bEnd.3 cells after 15 min of 5 μM of Aβ1–42 treatment in the presence of 5 μM of BAY 11-7082 compared to the cells that were treated with Aβ1–42 without BAY 11-7082, in which a significant induction was shown in comparison with the control cells (Figures 3f and g). In order to examine whether the NF-κB pathway mediated the decrease in P-gp expression, we used a NF-κB response element luciferase gene reporter assay, in which luciferase activity increases as NF-κB binds to its response element. Although Aβ1–42 alone significantly increased the NF-κB promoter luciferase activity, the co-treatment of 5 μM of Aβ1–42 with 5 μM of BAY 11-7082 for 30 min significantly attenuated the NF-κB promoter luciferase activity in bEnd.3 cells (Figure 3h). Together, these results indicated that NF-κB signaling mediated the Aβ1–42-induced decrease in P-gp expression.

The nuclear factor-kappa B (NF-κB) pathway mediated the Aβ1–42-induced decrease in P-gp expression. (a) Western blot analysis of the P-gp contents in bEnd.3 cells that were incubated with 5 μM of Aβ1–42 for 24 h with or without 5 μM of BAY 11-7082. (b) The histogram represents the densitometric analysis of the blots, with the data expressed as mean±S.E.M. (n=4; **P<0.01 versus Aβ 5 μM). (c) Confocal microscopic analysis of the P-gp intensity in the bEnd.3 cells that were treated with 5 μM of Aβ1–42 for 24 h with or without 5 μM of BAY 11-7082. The nuclei were labeled with DAPI (blue), and P-gp was immunofluorescently labeled in red. Scale bar=20 μm. (d) The fluorescence intensity profile of each imaged cell that was determined by drawing lines parallel to the axis of the cell. The x-axis shows the length of the line, which was ∼30 μm in total, and the y-axis shows the intensity. The red line shows the intensity of P-gp, and the blue line shows that of DAPI. (e) The average peak fluorescence intensity of P-gp is presented. The peak fluorescence intensity values were taken from 10 cells per group from three independent preparations (***P<0.001 versus Aβ 5 μM). (f) The p-IκBα protein levels were measured by a western blot analysis in bEnd.3 cells that were incubated with 5 μM of Aβ1–42 for 15 min with or without 5 μM BAY 11-7082, alongside total IκBα. (g) The histogram shows a densitometric analysis of the blots, with the data expressed as mean±S.E.M. (n=4; ***P<0.001 versus Aβ 5 μM). (h) NF-κB response element luciferase gene reporter assay done in bEnd.3 cells that were treated with Aβ1–42 for 30 min in the presence or absence of BAY 11-7082 (n=3; **P<0.01, ***P<0.001 versus Aβ 5 μM)
RAGE-mediated Aβ1–42-induced P-gp decrease
RAGE has been shown to activate multiple cellular signaling cascades, including the NF-κB pathway when its ligands, such as advanced glycation end product (AGE), S100, and Aβ, bind to it.29 The interactions of Aβ with RAGE at the BBB are involved in oxidative stress and in the activation of NF-κB.30 In order to determine whether RAGE mediated the Aβ1–42-induced decrease in P-gp expression, we used a neutralizing antibody against RAGE that blocks the Aβ1–42–RAGE interaction by recognizing the extracellular domain of RAGE.31 The anti-RAGE antibody attenuated the decrease in P-gp expression by Aβ1–42, as shown in the immunofluorescence analysis (Figure 4a). The representative fluorescence intensity profiles are shown in Figure 4b. The red-peak fluorescence intensity was decreased in Aβ1–42-treated bEnd.3 cells compared to that in control-treated bEnd.3 cells, and it increased when Aβ1–42 was cotreated with the anti-RAGE antibody in bEnd.3 cells. After Aβ1–42 treatment in the presence of 15 μg/ml of the anti-RAGE antibody, the average value of the red-peak fluorescence intensity was significantly rescued compared to that in Aβ1–42-treated bEnd.3 cells (Figure 4c), suggesting that RAGE plays a critical role in the P-gp reduction by Aβ1–42. The P-gp levels were measured in bEnd.3 cells that overexpressed the full-length human RAGE in the presence or absence of Aβ1–42 for 24 h. Aβ1–42 decreased the P-gp levels in RAGE-overexpressing bEnd.3 cells to a greater extent than mock-transfected cells (Figures 4d and e). These results suggested that RAGE mediates Aβ1–42-induced P-gp reductions through NF-κB signaling.

The receptor for advanced glycation end products (RAGE) mediated the Aβ1–42-induced decrease in P-gp expression. (a) Confocal microscopic analysis of the P-gp intensity in bEnd.3 cells that were treated with 5 μM of Aβ1–42 for 24 h with or without 15 μg/ml of anti-RAGE antibody. The nuclei were labeled with DAPI (blue), and the P-gp immunoreactivity was labeled in red. Scale bar=20 μm. (b) The fluorescence intensity profile of each imaged cell was determined by drawing lines that were parallel to the axis of the cell. The x-axis shows the length of the line, which was ∼30 μm in total, and the y-axis shows the intensity. The red line shows the intensity of P-gp, and the blue line shows that of DAPI. (c) The average peak fluorescence intensity of P-gp is presented. The peak fluorescence intensity values were taken from 10 cells per group, and from three independent preparations (***P<0.001 versus Aβ 5 μM). (d) P-gp expression levels were measured by western blots in bEnd.3 cells that were incubated with 5 μM of Aβ1–42 for 24 h after transfection with mock or full-length human RAGE. (e) The histogram represents the densitometric analysis of the blots, with the data expressed as mean±S.E.M. (n=3; **P<0.01, ***P<0.001 versus mock-ctrl)
The P-gp reductions by Aβ1–42 were attenuated when astrocytes contacted ECs
Along with other BBB properties, P-gp expression is induced when the cells are in contact with astrocytes in vitro and in vivo,13, 14 and this is primarily due to end-feet processes and some inducing factors that originated by themselves.32 Based on the findings of previous studies, in order to identify whether a contact or non-contact system between astrocytes and ECs affects the P-gp reduction by Aβ1–42, we compared the protein levels of P-gp by using the conditioned medium (CM) that was supplied from bEnd.3 cells that were grown in two different co-culture systems (Figure 5a). In one system, which was called the contact model, the primary cultured astrocytes were first seeded on the underside of the membrane of the collagen-coated Transwell inserts so that the astrocyte end-feet could make contact with the bEnd.3 cells that were later seeded in the insert on the upside of the membrane. In the other system, which was called the non-contact model, the astrocytes were seeded on the 24-well plate, and the bEnd.3 cells were later seeded inside the insert. For controls, bEnd.3 cells were cultured in the insert without the astrocyte feeder layer. The CM from the astrocytes-bEnd.3 cells of the co-culture plate for each system was transferred to the bEnd.3 cells, and the CM was changed daily until the Aβ1–42 treatment. Although the P-gp expression in bEnd.3 cells that were cultured in the CM in the non-contact model was similar to the P-gp protein levels in Aβ1–42-treated bEnd.3 cells, the Aβ1–42-induced decrease in P-gp expression was significantly attenuated in the presence of the CM of the contact model (Figures 5b and c). These results suggested that some unknown soluble factors that were released from the astrocytes making contact with the ECs could attenuate the Aβ1–42-induced decrease in P-gp expression.

Aβ1–42 induced a decrease in P-gp expression only when the astrocyte–endothelial cell (EC) contact was not made. (a) Schematic diagrams illustrating the conditioned medium (CM) transfer from the control, and the contact and non-contact bEnd.3-astrocyte co-culture systems. (b) Western blot analysis of the P-gp contents in bEnd.3 cells that were incubated with 5 μM of Aβ1–42 for 24 h in the presence of the described CM. (c) Histogram showing densitometric analysis of the blots, with the data expressed as mean±S.E.M. (n=4; **P<0.01 versus Aβ 5 μM)
Astrocyte polarization was disrupted in the brains of 5XFAD mice
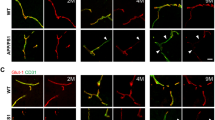
Because the in vitro results shown in Figure 5 strongly suggested that astrocyte–EC contacts were protective for the P-gp reduction by Aβ1–42, it was reasonable to examine change of astrocyte polarization in the brains of wild-type and 5XFAD mice. The capillaries were labeled with tomato lectin, which is an effective blood vessel marker. However, lectin is also a marker of microglia, and it labels the activated microglia around the amyloid plaques in 5XFAD mice with a different pattern compared to how it labels blood vessels, thus allowing us to predict where the amyloid plaque might be. Two markers for the polarized astrocyte end-feet, the end-feet specific channel aquaporin 4 (Aqp4) and the astrocyte-derived basement membrane component laminin α2 (Lama2), both provided uniform labeling of the capillary surfaces in the brains of wild-type mice (Figures 6a and c). However, in 5XFAD mice, the vascular staining of these markers, Aqp4 and Lama2, were significantly weaker than that in controls (Figures 6b and d). Aqp4 signals were diffused (white arrow heads) and astrocyte end-feet was localized to bridging microvessels, indicating a loss of astrocyte polarization. Together, these data suggested a loss of astrocyte polarization in 5XFAD mice, thus indicating the detachment of astrocyte end-feet from ECs in AD.

Astrocyte polarization was disrupted in the brains of 5XFAD mice. (a) Confocal microscopic analysis of aquaporin 4 (Aqp4) and blood vessels in the cortical sections (30 μm) from 6-month-old control and 5XFAD mice (n=3 for each). Aqp4 (red) was localized to the blood vessels (green) in controls, but was diffused (white arrow heads) and distributed to bridging microvessels in 5XFAD mice. Scale bar=50 μm. (b) Compared to wild-type mice, Aqp4-positive cells were significantly decreased in the brains of 5XFAD mice. (c) Confocal microscopic analysis of laminin α2 (Lama2) and the blood vessels in the cortical sections of 6-month-old control and 5XFAD mice (n=3 for each). Lama2 (red) was localized to the blood vessels (green) in controls, but it was reduced in intensity and in the extent of the covered area in 5XFAD mice. Scale bar=50 μm. (d) Compared to wild-type mice, Lama2-positive cells were significantly decreased in the brains of 5XFAD mice. Both P-values (*P<0.05 and ***P<0.001) means the 5XFAD mice versus the wild-type mice)
Discussion
The essential role of P-gp in protecting the brain from toxic substances, including Aβ,15, 16, 17, 19 in the brains of AD patients and animal models suggests that therapies that target P-gp or its regulatory factors might be promising for the treatment and prevention of the disease. However, the molecular mechanisms that explain how P-gp expression decreases under AD conditions have been largely unknown. Our in vivo data showed that P-gp expression was significantly reduced especially near amyloid plaques, and that the polarity of astrocytes was lost in 5XFAD AD model mice compared to wild-type mice. The results that were obtained from the murine brain EC line bEnd.3 showed that the RAGE–NF-κB signaling pathway mediated the Aβ-induced decrease in P-gp expression, and that the induction was possible only when ECs did not receive inductive signals or factors from astrocytes that were making contact with ECs. Based on these results, we propose a mechanism involving P-gp downregulation in AD brain ECs, by which Aβ induces NF-κB signaling through an interaction with RAGE in the ECs that have lost contact with astrocytes that can induce P-gp expression.
It has been reported that brain capillary membranes that are isolated from hAPP mice exhibit a marked decrease in P-gp.19 In addition, a study has shown a significant inverse correlation between P-gp expression and the deposition of Aβ in the brains of elderly, non-demented patients.17 Therefore, we planned to examine whether P-gp expression was also reduced in our 5XFAD AD model mice, and we found a pattern of P-gp expression, especially in relation to amyloid plaque deposition. 5XFAD mice are amyloid precursor protein/presenilin-1 (PS1) double-transgenic mice that coexpress five familial AD mutations that accelerate plaque development.20 To clearly observe the decrease in P-gp expression and its correlation with amyloid plaques, we used the isolated brain capillaries and brain slices of 6-month-old 5XFAD mice that already exhibited memory deficits and significant progression in the pathologies rather than early-stage AD model mice. Our finding of a selective loss of P-gp expression, especially near amyloid plaques, in the cerebral capillary endothelium of 5XFAD mice (Figure 1) suggested that Aβ might be the cause for the decrease in P-gp expression. As expected, P-gp expression was significantly reduced by Aβ in bEnd.3 cells after 24 h of treatment with 5 μM of Aβ1–42 (Figure 2).
Previous studies have shown that Aβ1–42 treatment reduces the mRNA expression of P-gp.23, 33 Knowing that Aβ reduces P-gp expression at the transcription level, we examined which signaling pathway mediated the Aβ-induced decrease in P-gp expression by using specific inhibitors. NF-κB is involved in one of the pathways that is related to cell survival and that has an important role in neurodegenerative disorders,34 and it is known to be activated in ECs by Aβ.35 Although NF-κB resides in the cytoplasm and is bound to its inhibitor molecule IκBα in resting cells, it is released upon activation from the phosphorylated inhibitor to translocate into the nucleus where it binds to the DNA κB sites to induce the activation or repression of specific genes.29, 34 BAY 11-7082 inhibits the NF-κB pathway by suppressing the phosphorylation of IκBα. In the presence of BAY 11-7082, the Aβ-induced decrease in P-gp expression and luciferase activity were both significantly attenuated (Figure 3), suggesting that NF-κB mediated the Aβ-induced decrease in P-gp expression by binding to the DNA to repress the P-gp gene.
When a ligand, such as Aβ, binds to RAGE, it has been shown that multiple cellular signaling cascades get activated, including the NF-κB pathway.36 In addition, RAGE itself is one of the genes that are activated by NF-κB, enabling RAGE to amplify.37 Aβ1–42 decreased the P-gp levels in RAGE-overexpressing bEnd.3 cells to a greater extent compared to that in mock-transfected cells (Figure 4), suggesting that RAGE mediated the Aβ1–42-induced decrease in P-gp expression. As Aβ accumulates, RAGE also increases, thus amplifying the signal even more. However, P-gp expression has been reported to decrease before the protein expression of RAGE begins to increase,19 suggesting that the RAGE increase contributes to the P-gp downregulation later on, but not in the early stages. Blocking the Aβ1–42–RAGE interactions with a neutralizing antibody against RAGE confirmed that RAGE mediates the Aβ-induced decrease of P-gp expression, as bEnd.3 cell treated with Aβ1–42 in the presence of anti-RAGE antibody showed attenuation (Figure 4).
The BBB is composed of ECs, astrocytes, a basement membrane, pericytes, and neurons that are in physical proximity to the endothelium.38 Among them, astrocytes tightly appose end-feet on the abluminal membrane of the brain capillaries in order to induce the BBB properties, including P-gp expression, with its end-feet processes and some other inducing factors.13, 32 In order to examine whether Aβ could also induce the P-gp reduction in the context in which ECs receive inductive signals or factors that are released from astrocytes that contact with ECs, we used the CM of the bEnd.3-astrocyte co-culture system. There have been studies that have compared P-gp expression in mono-cultured ECs with ECs that were cocultured with astrocytes that make contact through feet.13, 14 Our study differed in that we did not observe ECs that were actually making contact with astrocytes, but ECs that received the CM or the released factors of the contact or non-contact bEnd.3-astrocyte co-culture systems. We hypothesized that the secretome of the astrocytes that made contact with ECs would differ from that of astrocytes that were not in contact with ECs. We found that the released factors of the contact co-culture system rescued the Aβ1–42-induced decrease in P-gp expression, but not that of the non-contact co-culture system, suggesting that Aβ1–42 reduced the P-gp expression only when the astrocytes did not make contact with ECs. This finding may be particularly important from a pharmacological point of view, for it showed that there was a factor that was released from astrocytes that attenuated the Aβ-induced decrease in P-gp expression and that this factor should be found by a proteomic analysis of the secretome of astrocytes that make contact with ECs but not of that primary astrocytes that were cultured alone, as has been previously shown.39, 40 Further studies are needed to isolate and characterize the astrocyte-derived signaling molecule that attenuated the Aβ-induced decrease in P-gp expression.
In order to confirm that Aβ reduced the P-gp expression in ECs that were not in contact with astrocytes, we examined if markers for polarized astrocyte end-feet were actually weaker in 5XFAD mice compared to controls. There have been reports of astrocyte polarization loss from an early stage in mouse models of AD, and this was suggested by the swelling and detachment of astrocyte end-feet surrounding perivascular amyloid deposits41 and the redistribution of Aqp4 from end-feet membranes to non-end-feet membrane domains.42 Our in vivo data also showed that Aqp4 was diffused and distributed to bridging microvessels and that the vascular stainings for Aqp4 and Lama2 were weaker, indicating a loss of astrocyte polarization in 5XFAD mice compared to wild-type mice (Figure 6). Questions remain of how astrocytes lose polarization in AD or whether astrocyte polarization loss actually precedes the decrease of P-gp expression in vivo. However, the finding that astrocytes in 5XFAD lose polarization and detached from ECs confirmed that the endothelium of patients with AD is in an environment that is vulnerable to an Aβ-induced decrease in P-gp expression by not receiving the inductive factors that are released from astrocytes that are making contact with ECs.
Together, we would like to suggest a model of how P-gp expression decreases in AD. Normally, astrocytes tightly appose end-feet on the abluminal membrane of the brain capillaries in order to induce BBB properties, including P-gp expression.13, 32 In the brain of AD patients, astrocytes somehow lose its connection with ECs (Figure 6), and become disabled to induce P-gp expression. When inducing factors secreted by astrocytes that made contact with ECs were present, P-gp expression did not decrease although toxic Aβ were present. However, when those inducing factors were not present, Aβ could reduce the expression of P-gp (Figure 5). Our data collectively suggested that Aβ induced decrease in P-gp expression in ECs that lost contact with astrocytes through NF-κB signaling by interactions with RAGE. In addition, we suggest the high possibility that some factors that are released from astrocytes can attenuate the Aβ-induced decrease of P-gp expression. Given the critical involvement of P-gp in protecting the brain from toxic substances, including Aβ, determining the precise molecular mechanisms of the Aβ-induced decrease in P-gp expression and the existence of an unknown factor that could attenuate the effect, may provide therapeutic targets for treating AD.
Materials and Methods
Reagents
BAY 11-7082 was purchased from Merck Millipore (Darmstadt, Germany), the anti-RAGE antibody was purchased from R&D Systems, Inc. (Minneapolis, MN, USA), and the Aβ1–42 peptide was purchased from American Peptide Company Inc. (Sunnyvale, CA, USA) and was prepared as described.43 The following antibodies were used for immunodetection: anti-P-gp (Covance Inc., Princeton, NJ, USA) and anti-beta amyloid (6E10; Covance Inc.), anti-p-IκBα and anti-IκBα (Cell Signaling Technology, Inc., Beverly, MA, USA); anti-RAGE and anti-glucose transporter-1 (GLUT-1; EMD Millipore Corporation, Billerica, MA, USA), and anti-tubulin and anti-β-actin (Sigma-Aldrich Co., LLC, St. Louis, MO, USA).
Animals and tissue collection
Five familial AD mutations (5XFAD) mice (Tg6799; B6SJL-Tg [APPSwFlLon, PSEN*M146L*L286V] 6799Vas/J, stock no. 006554) that overexpressed three human APP 695 mutations (Swedish, Florida, and London) and two human PS1 mutations (M146L and L286V) of human PS1 under transcriptional control of the murine Thy-1 promoter were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in Seoul National University’s mouse facility. Founder transgenic mice were identified by polymerase chain reaction and non-transgenic littermate mice served as controls. The animal treatment and maintenance protocols were performed in accordance with the Principle of Laboratory Animal Care (NIH publication No. 85–23, revised 1985) and the Animal Care and Use Guidelines of Seoul National University, Seoul, Korea. All of the experiments were approved by the Institute of Laboratory Animal Resources of Seoul National University.
Brain capillary isolation
Brain capillaries were isolated from wild-type littermates (n=3) and 5XFAD mice (n=3) at 6 months of age as previously described19, 44 with minor modifications. In brief, 6-month-old mice were anesthetized with a mixture (3 : 1 ratio, 1 ml/kg, i.p.) of Zoletil 50 (Virbac, Carros, France) and Rompun (Bayer Korea, Seoul, Korea) and then decapitated. The brains were cleared of superficial large blood vessels, the choroid plexus and the meninges. The cerebral cortices were homogenized in 4 vol. of ice-cold phosphate-buffered saline (PBS). The homogenate was centrifuged at 3500 × g for 10 min. The pellets were rehomogenized in 4 vol. of PBS containing 15% Ficoll T-400 (Sigma-Aldrich Co., LLC). After centrifugation at 25 000 × g for 20 min, the myelin floating on top of the supernatant was removed. The brain capillaries that were separated as a pellet were then lysed.
Cell culture
The murine brain endothelial cell line bEnd.3 (ATCC, Manassas, VA, USA) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific Inc., Waltham, MA, USA) that was supplemented with 10% fetal bovine serum (Thermo Fisher Scientific Inc.), 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich Co., LLC) at 37°C in a humidified 5% CO2 incubator. Before the drug treatments, the media was replaced with DMEM.
Transfection
Transfection of the bEnd.3 cells was performed by Lipofectamine LTX (Life Technologies Corporation, Waltham, MA, USA) in accordance with the manufacturer’s protocols. The cells were seeded on 24-well plate (4 × 104 cells/cm2; SPL, Pocheon, Korea) or 12 well plate (7 × 104 cells/cm2; SPL Life Sciences Co., Ltd, Pocheon, Korea) for RAGE over expression, and on 12 well plate (4 × 104 cells/cm2; SPL Life Sciences Co., Ltd) for the NF-κB-response element luciferase reporter gene assay. The cDNA constructs were mixed with Lipofectamine LTX in Opti-MEM (Life Technologies Corporation). After 20 min of incubation, the mixture was added to the cell-culture medium.
Primary astrocyte culture
The primary astrocyte cultures were prepared as previously described45 with minor modifications. Briefly, the cortices of newborn (1 or 2-day old) ICR mice pups were aseptically removed, chopped into pieces and trypsinized to be plated on T75 culture flasks (Thermo Fisher Scientific Inc.). The primary astrocytes were maintained in DMEM that was supplemented with 10% FBS (HyClone, Irvine, CA, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in humidified 5% CO2 incubator. The astrocytes were detached by trypsinization and plated on poly-L-lysine-coated culture plates with supplemented DMEM.
Preparation of the in vitro BBB model
The in vitro BBB model was prepared as previously described.13, 14 Briefly, astrocytes were seeded (3 × 104 cells/cm2) on the bottom of collagen-coated Transwell inserts (surface area: 0.33 cm2, pore size: 0.4 μm, Corning Incorporated, Corning, NY, USA) or 24-well plates 2 days before the bEnd.3 cells were seeded (4 × 104 cells/cm2) inside the insert, on the upside of the membrane, and cultured to tight monolayers. The bEnd.3 cells were also plated on another 24-well plate (4 × 104 cells/cm2), while it was being seeded in the insert. The CM from the bEnd.3-astrocyte co-culture plate was transferred to the bEnd.3 plate every day to minimize the denaturation that occurs during conservation of the CM.46 After transferring the CM for the second time, the cells seeded on the co-culture plate were maintained in DMEM instead of supplemented DMEM. Before drug treatment on the bEnd.3 plate, CM from the co-culture plate was transferred for the last time. A schematic drawing of the bEnd.3-astrocyte co-culture model is depicted in Figure 5.
Western blot analysis
Isolated brain capillaries and bEnd.3 cells were lysed with RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, and 50 mM Tris-HCl, pH 7.4) containing protease inhibitors (Sigma-Aldrich Co., LLC) and phosphatase inhibitors (A.G. Scientific, Inc., San Diego, CA, USA). The protein extracts were quantified by a bicinchoninic protein assay (Thermo Fisher Scientific Inc.) and equally loaded on 7% or 10% glycine gels. The separated samples were transferred to a polyvinylidenedifluoride membrane and incubated with antibodies against the indicated proteins. The protein bands were visualized with a bioimaging analyzer (LAS-3000; Fujifilm Corporation, Tokyo, Japan) with enhanced chemiluminescence (ECL; GE Healthcare Life Sciences, Seoul, Korea). The images were captured and analyzed with a Multi-Gauge program (Fujifilm Corporation).
Luciferase reporter gene assay
After 24 h of drug treatment after the transfection, the cells were lysed by passive lysis buffer. The luciferase activities in the cell extracts were measured by a dual-luciferase reporter assay system (Promega Corporation, Madison, WI, USA).
Immunocytochemistry
bEnd.3 cells were seeded on 18-mm round coverslips. After drug treatment, the cells were washed twice with ice-cold PBS and fixed with 4% paraformaldehyde (PFA; BIOSESANG, Inc., Gyeonggi-do, Korea) in PBS for 20 min at room temperature (RT). After permeabilization with 0.5% Triton X-100 (Sigma-Aldrich Co., LLC) in PBS, the cells were incubated with anti-P-gp antibody (1 : 100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) overnight at 4°C. After PBS washes, the cells were incubated with fluorescent-labeled secondary antibody (1 : 500; Life Technologies Corporation) for 2 h at RT and counterstained with 4′-6-diamidino-2-phenylindole (DAPI) for 10 min.
Image analysis
All images were acquired with a confocal laser scanning microscope (FV10i-w; Olympus Corporation, Tokyo, Japan). The fluorescence intensity profile of each imaged cell was determined by drawing lines parallel to the axis of the cell.24 The numerical values of the peak fluorescence intensities along each line were used for analysis. For quantification of immunoreactivity of brain tissues, immunofluorescence images of the cerebral frontal cortex were taken and analyzed Lama2 and Aqp4-positive cells, respectively. The number of immunofluorescence-positive pixels in the cerebral frontal cortex areas from the acquired images was analyzed using the Image J processing software (National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry
Wild-type littermate (n=3) and 5XFAD (n=3) female mice at 6 months of age were anesthetized and perfused transcardially with PBS and 4% PFA in PBS and post-fixed in 4% PFA for 20 h. The brain slices were sectioned to a 30-μm thickness with a CM 1950 cryostat (Leica Microsystems GmbH, Nussloch, Germany). Free-floating sections of the frontal cortices, which contain plenty of capillaries, were pretreated with 70% formic acid for 20 min to retrieve the antigens and then incubated with the following primary antibodies: biotin-labeled 4G8 (1 : 700; Covance, Inc.), rabbit anti-P-gp (1 : 100; Santa Cruz Biotechnology, Inc.), Fluorescein Lycopersicon esculentum (Tomato) lectin (1 : 100; Vector Laboratories, Inc., Burlingame, CA, USA), rat anti-Lama2 (1 : 500; Abcam plc, Cambridge, MA, USA), and rabbit anti-Aqp4 (1 : 100; EMD Millipore Corporation) overnight at 4°C. Following extensive washes in PBS, the sections were incubated with secondary antibodies that were conjugated with Alexa Fluor dyes (1 : 500; Life Technologies Corporation) for 2 h at RT. Immunofluorescent images were taken with the aid of a confocal laser scanning microscope (FV10i-w; Olympus Corporation).
Three-dimensional-SIM image acquisition
To clarify the modification of P-gp expression by amyloid plaques, we used a super-resolution structured illumination microscopy (SIM; Nikon N-SIM, Nikon Instruments, Inc., Tokyo, Japan) as described.25 Briefly, images of the immunofluorescent stained brain slices were taken sequentially in the z-direction in order to reconstruct them into 3D-SIM images by NIS-E software (Nikon Instruments, Inc.).
Statistics
All data are presented as mean±standard error of the mean (S.E.M.). Unpaired t-tests (for 2 groups) or one-way ANOVA tests that were followed by Tukey’s multiple comparisons tests (for >2 groups) were executed for the statistical analyses (*P<0.05, **P<0.01, and ***P<0.001) with GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA).
Abbreviations
- BBB:
-
blood–brain barrier
- AD:
-
Alzheimer’s disease
- Aβ:
-
amyloid β-peptide
- 5XFAD:
-
five familial Alzheimer’s disease mutations
- RAGE:
-
receptor for advanced glycation end products
- NF-κB:
-
nuclear factor-kappa B
References
Braak H, Braak E . Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–259.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010; 330: 1774.
Weller RO, Subash M, Preston SD, Mazanti I, Carare RO . Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 2008; 18: 253–266.
Ohtsuki S, Terasaki T . Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res 2007; 24: 1745–1758.
Begley DJ, Brightman MW . Structural and functional aspects of the blood-brain barrier. Prog Drug Res 2003; 61: 39–78.
Neuwelt EA . Mechanisms of disease: the blood-brain barrier. Neurosurgery 2004; 54: 131–140 discussion 141–132.
Hawkins BT, Davis TP . The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 2005; 57: 173–185.
Wardlaw JM, Sandercock PA, Dennis MS, Starr J . Is breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke 2003; 34: 806–812.
Zlokovic BV . The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008; 57: 178–201.
Demeule M, Labelle M, Regina A, Berthelet F, Beliveau R . Isolation of endothelial cells from brain, lung, and kidney: expression of the multidrug resistance P-glycoprotein isoforms. Biochem Biophys Res Commun 2001; 281: 827–834.
Schinkel AH . The physiological function of drug-transporting P-glycoproteins. Semin Cancer Biol 1997; 8: 161–170.
Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H . Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia 1997; 19: 13–26.
Gaillard PJ, van der Sandt IC, Voorwinden LH, Vu D, Nielsen JL, de Boer AG et al. Astrocytes increase the functional expression of P-glycoprotein in an in vitro model of the blood-brain barrier. Pharm Res 2000; 17: 1198–1205.
Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ et al. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem 2001; 76: 1121–1128.
Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides—implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol 2007; 17: 347–353.
Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W et al. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 2002; 12: 535–541.
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 2005; 115: 3285–3290.
Hartz AM, Miller DS, Bauer B . Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol 2010; 77: 715–723.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26: 10129–10140.
Moon M, Hong HS, Nam DW, Baik SH, Song H, Kook SY et al. Intracellular amyloid-beta accumulation in calcium-binding protein-deficient neurons leads to amyloid-beta plaque formation in animal model of Alzheimer’s disease. J Alzheimers Dis 2012; 29: 615–628.
Kalaria RN . The blood-brain barrier and cerebrovascular pathology in Alzheimer’s disease. Ann N Y Acad Sci 1999; 893: 113–125.
Kania KD, Wijesuriya HC, Hladky SB, Barrand MA . Beta amyloid effects on expression of multidrug efflux transporters in brain endothelial cells. Brain Res 2011; 1418: 1–11.
Wijesuriya HC, Bullock JY, Faull RL, Hladky SB, Barrand MA . ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res 2010; 1358: 228–238.
Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I . Abeta(1)(−)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J Neurosci 2012; 32: 8845–8854.
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996; 382: 685–691.
Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P et al. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med 1995; 1: 693–699.
Ohkita M, Takaoka M, Shiota Y, Nojiri R, Sugii M, Matsumura Y . A nuclear factor-kappaB inhibitor BAY 11-7082 suppresses endothelin-1 production in cultured vascular endothelial cells. Jpn J Pharmacol 2002; 89: 81–84.
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 2005; 83: 876–886.
Deane R, Wu Z, Zlokovic BV . RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004; 35 (11 Suppl 1): 2628–2631.
Li M, Shang DS, Zhao WD, Tian L, Li B, Fang WG et al. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J Immunol 2009; 182: 5778–5788.
Kim JH, Kim JH, Park JA, Lee SW, Kim WJ, Yu YS et al. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol 2006; 39: 339–345.
Brenn A, Grube M, Peters M, Fischer A, Jedlitschky G, Kroemer HK et al. beta-Amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood-brain barrier in mice. Int J Alzheimers Dis 2011; 2011: 690121.
Mincheva-Tasheva S, Soler RM . NF-kappaB signaling pathways: role in nervous system physiology and pathology. Neuroscientist 2013; 19: 175–194.
Gonzalez-Velasquez F, Reed JW, Fuseler JW, Matherly EE, Kotarek JA, Soto-Ortega DD et al. Activation of brain endothelium by soluble aggregates of the amyloid-beta protein involves nuclear factor-kappaB. Curr Alzheimer Res 2011; 8: 81–94.
Bierhaus A, Humpert PM, Stern DM, Arnold B, Nawroth PP . Advanced glycation end product receptor-mediated cellular dysfunction. Ann N Y Acad Sci 2005; 1043: 676–680.
Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H . The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem 2000; 275: 25781–25790.
Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD . Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 2006; 1: 223–236.
Lafon-Cazal M, Adjali O, Galeotti N, Poncet J, Jouin P, Homburger V et al. Proteomic analysis of astrocytic secretion in the mouse. Comparison with the cerebrospinal fluid proteome. J Biol Chem 2003; 278: 24438–24448.
Skalnikova H, Motlik J, Gadher SJ, Kovarova H . Mapping of the secretome of primary isolates of mammalian cells, stem cells and derived cell lines. Proteomics 2011; 11: 691–708.
Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM . Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol 2011; 122: 293–311.
Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LM, Nilsson LN et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J Alzheimers Dis 2011; 27: 711–722.
Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ . Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 2002; 277: 32046–32053.
Dallaire L, Tremblay L, Beliveau R . Purification and characterization of metabolically active capillaries of the blood-brain barrier. Biochem J 1991; 276 (Pt 3): 745–752.
Benavides A, Pastor D, Santos P, Tranque P, Calvo S . CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia 2005; 52: 261–275.
Kuchler-Bopp S, Delaunoy JP, Artault JC, Zaepfel M, Dietrich JB . Astrocytes induce several blood-brain barrier properties in non-neural endothelial cells. Neuroreport 1999; 10: 1347–1353.
Acknowledgements
This work was supported by grants from the National Research Foundation (2012R1A2A1A01002881, 2013M3C7A1069644, and the Medical Research Center (2011-0030738)); the Korea National Institute of Health ROAD R&D Program Project (A092058), and the KIST Institutional Program (2E24242-13-135) for IM-J.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Park, R., Kook, SY., Park, JC. et al. Aβ1–42 reduces P-glycoprotein in the blood–brain barrier through RAGE–NF-κB signaling. Cell Death Dis 5, e1299 (2014). https://doi.org/10.1038/cddis.2014.258
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.258
This article is cited by
-
Role of Brain Liver X Receptor in Parkinson’s Disease: Hidden Treasure and Emerging Opportunities
Molecular Neurobiology (2024)
-
Effect of Apolipoprotein E isoforms on the Abundance and Function of P-glycoprotein in Human Brain Microvascular Endothelial Cells
Pharmaceutical Research (2024)
-
Loss of Direct Vascular Contact to Astrocytes in the Hippocampus as an Initial Event in Alzheimer’s Disease. Evidence from Patients, In Vivo and In Vitro Experimental Models
Molecular Neurobiology (2024)
-
SIRT1 pathway in Parkinson’s disease: a faraway snapshot but so close
Inflammopharmacology (2023)
-
Parkinson’s Disease Risk and Hyperhomocysteinemia: The Possible Link
Cellular and Molecular Neurobiology (2023)
