
Abstract
Adaptive responses of skeletal muscle regulate the nuclear shuttling of the sarcomeric protein Ankrd2 that can transduce different stimuli into specific adaptations by interacting with both structural and regulatory proteins. In a genome-wide expression study on Ankrd2-knockout or -overexpressing primary proliferating or differentiating myoblasts, we found an inverse correlation between Ankrd2 levels and the expression of proinflammatory genes and identified Ankrd2 as a potent repressor of inflammatory responses through direct interaction with the NF-κB repressor subunit p50. In particular, we identified Gsk3β as a novel direct target of the p50/Ankrd2 repressosome dimer and found that the recruitment of p50 by Ankrd2 is dependent on Akt2-mediated phosphorylation of Ankrd2 upon oxidative stress during myogenic differentiation. Surprisingly, the absence of Ankrd2 in slow muscle negatively affected the expression of cytokines and key calcineurin-dependent genes associated with the slow-twitch muscle program. Thus, our findings support a model in which alterations in Ankrd2 protein and phosphorylation levels modulate the balance between physiological and pathological inflammatory responses in muscle.
Similar content being viewed by others


Main
Ankyrin repeat domain protein 2 (Ankrd2/Arpp) belongs to the muscle ankyrin repeat protein (MARP) family, whose members are involved in muscle stress response pathways.1 In addition to a well-documented role of Ankrd2 in mechanotransduction,2, 3, 4, 5, 6 we previously demonstrated a role of Ankrd2 in coordinating proliferation and apoptosis during myogenic differentiation,7 which we found to be dependent on Akt2-mediated phosphorylation at Ser-99 in response to oxidative stress.8 Although the binding of Ankrd2 to numerous nuclear and cytoplasmic proteins9 suggests its involvement in various signaling pathways, its functional significance has remained elusive. Based on ablation and overexpression of Ankrd2 in differentiating primary myoblasts and C2C12 cells, we demonstrate that Ankrd2 plays an important role in modulating NF-κB transcriptional activity through direct interaction with the NF-κB repressor p50. NF-κB is a transcription factor composed of homo- and heterodimers, affecting the expression of more than 150 genes related to inflammatory and cell survival responses.10 Tight regulation of NF-κB target genes is essential for ensuring the appropriate induction and subsequent resolution of inflammation and fluctuations in the levels of NF-κB-dependent cytokines establish whether the effects of inflammatory signaling pathways are beneficial or detrimental.11 Rapid resolution of inflammation is ensured by the NF-κB-specific inhibitor IκBα.12 Furthermore, several additional regulatory checkpoints regulate NF-κB activity. In particular, the p50 subunit was recently shown to play a predominant role in the suppression of inflammation during the later stages of NF-κB activation through the formation of specific NF-κB complexes.13 As p50 lacks a transactivation domain, its influence on transcription is dependent on dimerization with other members of the NF-κB family or on different coregulators.13, 14, 15 Here we show that Ankrd2 recruits p50, dominating over p65/p50 dimers, and orchestrates the repression of inflammation-related genes during muscle differentiation. As a direct target of the novel p50/Ankrd2 dimer, we have identified Gsk3β that has emerged as an important player for both generation and resolution of inflammatory responses.16, 17, 18 Another important regulator of NF-κB function is the intracellular redox state. Reactive oxygen species (ROS) activate NF-κB and enhance the expression of pro-inflammatory genes, such as TNFα, IL6, and C-reactive protein, which are involved in the pathogenesis of inflammation.19 Pro-inflammatory cytokines, in turn, stimulate the production of ROS that have thus been proposed as universal second messengers of the NF-κB activation pathway.20 Cells possess several defense systems to protect against the harmful effects of ROS. Here we propose a novel molecular mechanism by which activation of Akt2 leads to the resolution of oxidative stress and inflammation via Ankrd2-dependent NF-κB inhibition. Unexpectedly, the absence of Ankrd2 in slow muscle in vivo was associated with decreased expression levels of cytokines and key calcineurin-dependent genes, involved in the maintenance of the slow-twitch muscle program. Thus, as muscles produce cytokines both constitutively and in response to various inflammatory stimuli,21, 22 our findings suggest a multifaceted role of Ankrd2 in inflammatory signaling pathways.
Results
Inverse correlation between Ankrd2 levels and the expression of pro-inflammatory genes
To study the role of Ankrd2, we took advantage of the Ankrd2 knockout (KO) mouse model (Supplementary Figure S1)23 and performed global transcriptome analysis on proliferating, fusing, and differentiated primary myoblasts derived from wild-type (WT) and KO mice infected with adenovirus expressing HA-tagged Ankrd2 (AdAnkrd2) or control GFP (AdGFP). Quantitative real-time PCR (qRT-PCR) and western blot analyses showed highly efficient gene transfer of Ankrd2 (Supplementary Figures S2a and b). Gene expression profiles of differentiating myoblasts after deletion, rescue, or overexpression of Ankrd2 were obtained as outlined in Supplementary Figure S2c and the complete lists of altered genes can be found in Supplementary Datasets S1, S2, and S3, respectively. As shown in Figures 1a and b and in Supplementary Table S1, the most affected gene network was associated with inflammatory and immune responses. As Ankrd2 is expressed at increasing levels during differentiation, the major transcriptional changes were found in Ankrd2 KO myotubes, where the expression of many crucial NF-κB-inducible genes, including the pro-inflammatory cytokines Tnfα, Il1β, and Il6, was increased (see Supplementary Dataset S1). Conversely, these genes were downregulated by Ankrd2 overexpression (see Supplementary Datasets S2 and S3), which affected the expression of 69 transcripts related to inflammation, having NF-κB as a central node (Figure 1c). Consistent with a role of Ankrd2 as a negative regulator of NF-κB-directed inflammation, a specific enrichment for NF-κB-binding motifs was found in the set of downregulated genes in Figure 1c using the oPOSSUM tool (Figure 1d).

Ankrd2 protein levels affect the expression of NF-κB-related genes. (a) KEGG pathway enrichment analysis for up- and down-regulated genes in fully differentiated myoblasts from AdGFP-infected Ankrd2 KO cells compared with WT. A total of 2291 differentially genes (1381 upregulated and 910 downregulated) were identified by unpaired two-class significance analysis of microarray (SAM) analysis with a false discovery rate (FDR) of 0%. Only selected pathways with the most significant changes are shown (P<0.1). (b) KEGG pathway enrichment analysis for up- and downregulated genes in AdAnkrd2- compared with control AdGFP-infected Ankrd2 KO cells at different stages. Three sets of unpaired two-class SAM analyses showed that Ankrd2 overexpression resulted in differential expression of a total of 48, 197, and 235 unique transcripts in proliferating, fusing, and differentiated Ankrd2 KO myoblasts, respectively (0% FDR). (c) Gene expression heat map for the functional annotated transcripts that are differentially expressed (0% FDR) at all differentiation stages in response to acute overexpression of Ankrd2 in Ankrd2 KO cells and the relative pie chart showing the KEGG pathway enrichment analysis. The value in each sector represents the number of deregulated genes. (d) Identification of transcription factor binding sites (TFBSs) present in the DE genes by oPOSSUM, showing a significant enrichment for NF-κB-binding sites
Ankrd2 inhibits NF-κB transcriptional activity
To determine the effect of Ankrd2 on NF-κB activity, myogenic C2C12 cells were cotransfected with a NF-κB reporter vector containing a triplicate NF-κB-responsive element driving a firefly luciferase reporter (3 × NF-κB-luc) and Renilla luciferase vector (pGL4.74) as normalization control before infection with AdAnkrd2 or control AdGFP. NF-κB activity increased during myogenic differentiation, but was repressed by Ankrd2 at all stages (Figure 2a), identifying Ankrd2 as a negative regulator of NF-κB. We recently demonstrated that under hydrogen peroxide-induced stress triggering the production of ROS, Akt2 phosphorylates Ankrd2 at Ser-99, inducing its nuclear translocation and inhibition of muscle differentiation.8 To determine whether Akt2-dependent Ankrd2 phosphorylation mediates NF-κB regulation, C2C12 myoblasts were cotransfected with the 3 × NF-κB-luc reporter vector and FLAG-tagged WT or Ser99Ala mutant human Ankrd2. As shown in Figure 2b, WT Ankrd2, but not nonphosphorylatable Ser99Ala-Ankrd2, inhibited NF-κB activity both during proliferation and differentiation, demonstrating that Akt can inhibit NF-κB-regulated inflammatory genes via phosphorylation of Ankrd2 and suggesting a role of Ankrd2 as a sensor of the oxidative state through impairment of NF-κB activation. To determine whether Akt exerts an inhibitory effect on NF-κB in a ROS- and Ankrd2-dependent manner during muscle differentiation, we studied the effect of hydrogen peroxide treatment and found increased phosphorylation levels of Akt and Ankrd2 and consequent decreased NF-κB luciferase activity (Figures 2c and d). As expected, inhibition of Akt led to dephosphorylation of Ankrd2, but a decrease in NF-κB activity, consistent with the well-established role of Akt in promoting NF-κB transcriptional activity.24, 25 Our results, revealing a novel mechanism by which Akt-dependent Ankrd2 phosphorylation negatively regulates NF-κB during oxidative stress, suggest for the first time that Akt can act both positively and negatively on the NF-κB pathway.

Ankrd2 represses the transcriptional activity of NF-κB during oxidative stress. (a) C2C12 myoblasts were transfected with the 3 × NF-κB-luc vector and pGL4.74 control vector before infection with AdAnkrd2 or AdGFP. The values represent the firefly/Renilla luciferase ratio in proliferating and differentiating cells. (b) Luciferase activity assay in C2C12 cells following cotransfection of 3 × NF-κB-luc vector with FLAG-tagged WT or Ser99Ala mutant (S99A) Ankrd2. (c) Effect of 4 h of treatment with hydrogen peroxide on NF-κB transcriptional activity in C2C12 myoblasts at 1 day after induction of differentiation. Where indicated, cells were pretreated with Akt1/2- and Akt-specific inhibitors, as previously described.8 (d) Western blot analyses on the same samples as in (c), showing increased phosphorylation levels of Akt and Ankrd2. MB, myoblast; MT, differentiating myotubes at 1, 3, and 6 days (d) after induction. Data are represented as mean±S.D. (n=3). *P<0.05; **P<0.01
Ankrd2 interacts and colocalizes with the NF-κB member p50
Based on the striking similarity between Ankrd2 and the NF-κB inhibitor I-κB,26 we hypothesized that Ankrd2 may modulate NF-κB activity through direct interaction. To test the ability of Ankrd2 to bind to the two main subunits of NF-κB, p65 or p50, we transfected C2C12 cells with FLAG-tagged human WT Ankrd2 and performed immunoprecipitation (IP) using anti-FLAG-tag antibody, followed by western blot analysis for p50 and p65. This revealed the binding of Ankrd2 to p50, but not p65 (Figure 3a), which was confirmed by a reverse experiment, showing the ability of p50 to immunoprecipitate Ankrd2 (Figure 3b). FLAG-tagged Ser99Ala-mutant Ankrd2 failed to co-immunoprecipitate p50, demonstrating that phosphorylation of Ser-99 is critical for the Ankrd2–p50 interaction.

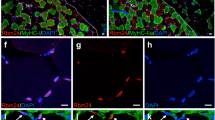
Ankrd2 interacts and colocalizes with p50. (a) IP of Ankrd2 following transfection of C2C12 cells with FLAG-tagged WT or Ser99Ala mutant Ankrd2. Ankrd2 is able to interact with p50, but not with p65. Moreover, Ankrd2 Ser99Ala does not bind to p50, demonstrating that Ankrd2 phosphorylation is essential for p50 binding. (b) Reverse experiment confirming the interaction between Ankrd2 and p50: IP of p50 following transfection of C2C12 cells with FLAG-tagged p50. (c) Immunostainings of proliferating and differentiating C2C12 cells for Ankrd2 and p50. Both proteins were visible in a speckled pattern in the nuclei of proliferating and early differentiating myoblasts (panel I at two different magnifications and panel II). With progressive differentiation, Ankrd2 became almost completely cytoplasmic and did not colocalize with p50 (panels III and IV at two different magnifications). MB, proliferating myoblasts, MT, differentiating myotubes at 1, 3, and 6 days (d) after induction
To identify the cellular compartment where p50 interacts with Ankrd2, we investigated the localization of endogenous Ankrd2 during proliferation and differentiation. In proliferating and early differentiating C2C12 cells, colocalization of Ankrd2 and p50 in structures resembling promyelocytic leukemia (PML) nuclear bodies (NBs)27 was found (Figure 3c, panels I and II). However, upon advancing differentiation, Ankrd2 moved to the cytoplasm and colocalization was lost, except in few cells where Ankrd2 was still present in the NBs (Figure 3c, panels III and IV). As we observed repression of NF-κB by exogenous Ankrd2 protein in terminally differentiated myotubes where endogenous Ankrd2 and p50 do not colocalize, we stained AdAnkrd2-infected C2C12 myotubes with anti-HA antibody to detect exogenous Ankrd2. As shown in Supplementary Figure S3, when overexpressed, Ankrd2 translocates to the nucleus where it may interact with p50 and inhibit NF-κB activity, thereby regulating inflammation-related genes.
Gsk3β is transcriptionally regulated by the Ankrd2/p50 complex
To validate the Ankrd2/p50 complex as a novel repressor, we searched for putative targets among genes whose expression levels were anticorrelated with that of Ankrd2. Interestingly, the only gene that was strongly downregulated upon overexpression of Ankrd2 at all stages was Gsk3β, a powerful governor of inflammatory signaling (Figure 4a). Western blot analysis confirmed the decrease in GSK3β protein and phosphorylation levels (Figure 4b), suggesting that the inhibition of NF-κB signaling by Ankrd2 overexpression may be reinforced through the Gsk3β pathway.28 These results were confirmed in C2C12 cells where infection with AdAnkrd2 resulted in decreased phosphorylation of Gsk3β at all stages (Figure 4c), whereas the protein level was significantly reduced only in fully differentiated cells (Figure 4c). Overexpression of Ser99Ala-mutant Ankrd2 had no effect on Gsk3β phosphorylation (Figure 4d), demonstrating that the effect of Ankrd2 on Gsk3β activity is dependent on its phosphorylation by Akt2. These results identify Gsk3β as a downstream target of the anti-inflammatory signaling cascade activated by Akt2-phosphorylated Ankrd2.

Ankrd2 decreases the Gsk3β protein expression level and inhibits its activity. (a) Heat map of gene expression values for Ankrd2 and Gsk3β in proliferating (stage 1), fusing (stage 2), and differentiated (stage 3) primary myoblasts derived from Ankrd2 KO and WT mice infected with AdAnkrd2 or control AdGFP (n=3). (b) Representative western blot showing Gsk3β protein and phosphorylation levels at the same stages as in (a). (c) Western blot analysis for Ankrd2 and Gsk3β protein and phosphorylation levels on lysate from proliferating and differentiating C2C12 myoblasts infected with AdAnkrd2 or AdGFP. Densitometry analysis for total and phosphorylated Gsk3β normalized to β-actin protein levels was performed. (d) Western blot and relative densitometry analysis of active and total Gsk3β normalized to β-actin protein levels from proliferating and differentiating C2C12 cells transfected or not with WT or Ser99Ala mutant (S99A) Ankrd2. MB, proliferating myoblasts, MT, differentiating myotubes at 1 and 6 days (d) after induction. Data are represented as mean±S.D. (n=3). *P<0.05; **P<0.01
Using the Genomatix and Patch tools, we identified several putative binding sites for NF-κB transcription factors in the Gsk3β upstream promoter region (Figure 5a). To determine whether Ankrd2 and NF-κB are recruited to the Gsk3β promoter, chromatin immunoprecipitation (ChIP) was performed on proliferating and differentiated C2C12 cells using antibodies against Ankrd2, p50, or p65, after which the captured genomic DNA was analyzed by qRT-PCR using primers spanning the Gsk3β promoter. As shown in Figures 5b and c, specific binding sites for Ankrd2 and p50 were found within the Gsk3β promoter. To functionally validate these interactions, C2C12 cells were cotransfected with a luciferase reporter construct containing selected Ankrd2- and p50-binding sites within the Gsk3β promoter and expression vectors encoding FLAG-tagged WT or Ser99Ala mutant human Ankrd2 or empty expression plasmid as a negative control. Cotransfection with WT Ankrd2 caused significant repression of Gsk3β promoter activity, whereas mutant Ankrd2 resulted in a much lower repression (Figure 5d). Thus, the complex formed by p50 and phosphorylated Ankrd2 can repress Gsk3β through direct binding to its promoter in an Akt2-phosphorylation-dependent manner.

Ankrd2 regulates the transcription of Gsk3β. (a) Schematic representation of putative NF-κB-binding elements within the Gsk3β promoter identified using Genomatix and Patch software. The white bars depict the regions of the Gsk3β promoter amplified by primer sets covering the putative NF-κB response elements. The Gsk3β luciferase reporter construct is represented on the bottom. (b) Using anti-Ankrd2 antibody, ChIP was performed on nuclear extracts from proliferating and differentiating C2C12 myoblasts. qRT-PCR was performed on input and immunoprecipitated DNA with the Gsk3β promoter-specific primers shown in (a). The bars represent the fold enrichment of Ankrd2 occupancy of the Gsk3β promoter relative to that of control IgG samples. (c) ChIP was performed using anti-p50 and anti-p65 antibodies followed by qRT-PCR analysis as described in (b). (d) Luciferase activity assay following cotransfection of C2C12 cells with a luciferase reporter plasmid encoding the identified Ankrd2 and p50-binding region within the Gsk3β promoter (765 bp, (a)) and WT or S99A mutant Ankrd2. pGL4.74 was used for normalization of transfection efficiency. MB, proliferating myoblasts, MT, differentiating myotubes at 1, 5, and 6 days (d) after induction. All data are represented as mean±S.D. (n=3). *P<0.05; **P<0.01
p50 regulates the expression of Ankrd2 in a classic feedback inhibition way
Ankrd2 was recently shown to be activated by longitudinal stretch through the recruitment of the NF-κB heterodimer p50/p65 to the Ankrd2 promoter that contains two putative NF-κB-binding sites.29 To investigate whether the NF-κB boxes are functional, we performed ChIP on proliferating and fully differentiated C2C12 myoblasts using antibodies against p50 and p65. The relative occupancy of the boxes (referred to as proximal and distal NF-κB box according to their position relative to the transcription start site) was determined by qRT-PCR using specific primers encompassing the NF-κB-binding sites. Our results showed strong binding of p50 to both the proximal and distal NF-κB boxes in myoblasts, whereas in differentiated cells, reduced binding to the proximal box and a complete abrogation of binding to the distal box was found (Figure 6a). Interestingly, only weak binding of p65 to the NF-κB boxes in the Ankrd2 promoter was detected in both myoblasts and differentiated cells, again with a reduction to baseline for the distal box in myotubes.

Ankrd2 expression is negatively and positively regulated by p50 and MyoD, respectively. (a) ChIP with antibodies against p50 and p65 was performed on nuclear extracts from proliferating and differentiating C2C12 cells followed by qRT-PCR using primers spanning the putative NF-κB-binding elements. The bars represent the fold enrichment of p50 and p65 occupancy of the Ankrd2 promoter relative to that of control IgG samples. (b) Graphical representation of the 5′-mouse Ankrd2 promoter region and the conserved portions in the constructs used for transfection experiments. (c) Luciferase activity assay following cotransfection of C2C12 cells with the first four luciferase constructs in (b) and the pGL4.74 control vector. (d and f) Luciferase activity assay in LPS-treated (2 μg/ml LPS for 24 h) compared with untreated cells transfected with the reporter vectors in (b). (e) Following cotransfection of HEK293A cells with FLAG-tagged MyoD and Ankrd2 promoter-driven luciferase vectors, ChIP was performed using anti-FLAG antibody. The immunoprecipitates were analyzed by standard PCR (top) and qRT-PCR (bottom) using specific primers for amplifying regions within the Ankrd2 promoter region containing the proximal or distal MyoD-binding site or both. IPed chromatin from HEK293A cells transfected with FLAG-tagged empty vector (IP mock) represents the negative control. The enrichment of each specific region of the Ankrd2 promoter compared with mock IP DNA was determined by qRT-PCR. All data are represented as mean±S.D. (n=3). MB, proliferating myoblasts, MT, differentiating myotubes at 1, 2, and 5 days (d) after induction. *P<0.05; **P<0.01
To investigate the functionality of the two NF-κB boxes within the Ankrd2 promoter, we performed a series of luciferase assays with the constructs shown in Figure 6b. The shortPROwtluc vector contains 317 bp upstream of the Ankrd2 ATG start codon spanning two paired E-boxes and the proximal NF-κB box. The longPROwtluc construct (−663 bp upstream of ATG) also includes the distal NF-κB box. Mutant reporters with deletion of the proximal box (shortPRO-NF-κBluc and longPRO-NF-κBluc) were obtained by site-directed mutagenesis. The reporter gene activity for each construct was measured in both proliferating and differentiated myoblasts as summarized in Figure 6c. Consistent with our previous data,7 both WT constructs resulted in increased luciferase activity and thus increased Ankrd2 expression during differentiation. On the other hand, activation of luciferase activity by the shortPROwtluc vector was about three times stronger than by the longPROwtluc construct, suggesting that a transcription factor may bind to the distal region of the Ankrd2 promoter negatively regulating its expression. Bioinformatic analysis suggested NF-κB as a repressor of the Ankrd2 promoter,29 which is consistent with our finding that p50 can directly bind to the Ankrd2 promoter and the fact that p50 lacks a transactivation domain and thus is mainly associated with transcriptional repression.30 These data, together with our finding that deletion of the proximal NF-κB regulatory element strongly increases Ankrd2-driven luciferase expression, suggest that the p50/p50 homodimer acts as a transcriptional repressor of Ankrd2 expression by binding its promoter.
It is well known that treatment of cells with proinflammatory cytokines (TNFα and IL1β), or bacterial lipopolysaccharides (LPS), leads to degradation of I-κB, allowing p50/p65 dimers to translocate to the nucleus and act as transcription factors.31 Consistent with this, activation of NF-κB by LPS was confirmed by an approximately twofold increase in luciferase activity in LPS-treated proliferating and differentiating C2C12 cells transfected with the NF-κB reporter vector (Supplementary Figure S4). Therefore, to study the effect of NF-κB activation on Ankrd2 expression, proliferating and differentiating C2C12 myoblasts transfected with the different luciferase constructs were treated with LPS. As shown in Figure 6d, luciferase activity was repressed following LPS treatment independent from the deletion of the proximal NF-κB box, suggesting that the p50/p65 dimer is not involved in the regulation of Ankrd2 expression. We have previously shown that Ankrd2 expression is under the control of MyoD,32 which is downregulated in response to TNFα-induced NF-κB activation.33 Consistently, a ChIP assay revealed direct binding of MyoD to the Ankrd2 promoter (Figure 6e). Moreover, transfection with the shortPRO-MyoDluc vector in which the MyoD-binding site was deleted resulted in a 2.7-fold reduction in luciferase activity as compared with the WT construct (Figure 6f), demonstrating that the Ankrd2 promoter is positively regulated by MyoD. Thus, the inhibition of the Ankrd2 promoter following LPS treatment during myoblast differentiation may be because of degradation of the Ankrd2 transcriptional activator MyoD at both the mRNA and protein levels, induced by activation of the canonical NF-κB pathway.33 This hypothesis is supported by the fact that the luciferase activity of the shortPRO-MyoDluc vector was not repressed following LPS treatment (Figure 6f).
Ankrd2 affects the expression of inflammatory cytokines in vivo
Our experiments on differentiating mouse primary and C2C12 myoblasts demonstrate a complex role of Ankrd2 in the pathways modulating inflammatory responses. To determine whether Ankrd2 may affect cytokine expression in vivo, mRNA and protein levels of NF-κB-regulated genes were determined by qRT-PCR and western blot analyses on slow oxidative soleus and fast glycolytic extensor digitorum longus (EDL) muscles from WT and Ankrd2 KO mice. Consistent with the previously reported preferential expression of Ankrd2 in slow-twitch fibers,34 Ankrd2 was expressed at higher amounts in soleus compared with EDL muscle (Figure 7a and Supplementary Figure S5). Furthermore, knockout of Ankrd2 was not compensated for by upregulation of other MARP family members (Figure 7a). Unexpectedly, decreased transcript levels of Il6, Tnfα, and IκBα were found in soleus muscle in the absence of Ankrd2 (Figure 7b), although Akt, Gsk3β, and NF-κB activities were similar between WT and Ankrd2 KO mice (Supplementary Figure S5). As NF-κB activity is physiologically higher in slow than in fast muscle35 (Supplementary Figure S5), we investigated whether Ankrd2 deletion may make muscle prone to fiber-type switching by changing the muscle transcriptional signature. As shown in Figure 7c, although skeletal muscle from Ankrd2 KO mice appeared morphologically equal to WT muscle and no significant changes in fiber-type distribution were found,23 the slow gene expression program was significantly modified. In particular, the calcineurin-dependent genes, Il6, Glut4, and Rcan1,36, 37, 38 were significantly downregulated in soleus muscle from Ankrd2 KO mice. Thus, our in vivo results suggest a role of Ankrd2 in the regulation of basal inflammatory cytokine production in a fiber type-dependent manner.

Ankrd2 affects the expression of inflammatory genes in a fiber-type specific manner in vivo. qRT-PCR analysis of (a) MARP family members, (b) NF-κB-dependent, and (c) calcineurin-responsive genes in slow oxidative soleus and fast glycolytic EDL muscle from 3-month-old WT and Ankrd2 KO mice. *P<0.05; **P<0.01
Discussion
Ankrd2 is strongly and rapidly upregulated under different muscle stress conditions, but the reason for this response has remained unclear. To explore gene networks that are disturbed by changes in Ankrd2 levels, we performed genome-wide expression analyses on proliferating and differentiating primary myoblasts derived from WT and Ankrd2 KO mice infected with adenovirus overexpressing Ankrd2 or control GFP. We chose to perform our study on primary mouse myoblasts, as gene expression analysis on whole muscle with nonhomogenous fiber composition is likely to show only an averaged picture of the consequences of Ankrd2 loss because of its fiber type-specific expression.33 Our analyses revealed that Ankrd2 negatively regulates the expression of a large number of genes known to be involved in inflammatory pathways mediated by NF-κB. NF-κB has been shown to control myogenesis through its involvement in both pro- and anti-myogenic pathways,39 and we here uncover a novel NF-κB signaling axis involved in muscle differentiation. More specifically, we establish Ankrd2 as an inhibitor of NF-κB activity through its interaction with the repressive NF-κB subunit p50 and inhibition of the expression of crucial inflammation-related genes. This is consistent with its striking sequence and structural similarity to I-κB, the specific NF-κB inhibitor.26 Similarly, overexpression of the MARP protein family member Ankrd1 resulted in decreased DNA binding of the NF-κB subunit p65 through an unknown mechanism.40 p50 can operate either as a pro-inflammatory or a negative regulator of NF-κB-directed inflammation depending on the cofactors that interact with the complex on the DNA. Our results reveal a role of Ankrd2 as a selective regulator of p50/p50 NF-κB-responsive genes in muscle cells. In particular, we found colocalization of Ankrd2 and p50 in a speckled intranuclear pattern, consistent with the presence of Ankrd2 in PML-NB multiprotein nuclear structures9 where transcriptional factors (i.e., NF-κB) and cofactors (i.e., Ankrd2) associate to regulate gene expression.27 This is interesting as PML has been shown to repress the transactivating function of NF-κB by recruiting p65 to PML-NBs and to interfere with the binding of NF-κB to its cognate DNA recognition sequence.41 In addition to upregulation of NF-κB target genes, Pparg and other members of the PPAR pathway were found to be downregulated in Ankrd2 KO cells. Activated PPARs can exert anti-inflammatory activity in several cell types, including skeletal muscle cells. In particular, PPARγ was recently shown to suppress cytokine-induced NF-κB transcriptional activity and target gene expression independent of p65 nuclear translocation and DNA binding.42 Thus, our findings suggest that PPAR-dependent inhibition of NF-κB activity may be mediated through the recruitment of Ankrd2 as a transcriptional cofactor of the NF-κB DNA complex.
As a target of the Ankrd2/p50 repressor complex, we identified Gsk3β, a crucial regulator of inflammatory processes.43 Thus, Ankrd2 reinforces its anti-inflammatory protection by negatively regulating the expression of Gsk3β, which is required for the production of pro-inflammatory cytokines, such as IL6, IL1ß, and TNFα.44 Consistently, Gsk3β, Il6, Il1β, and Tnfα were all downregulated after Ankrd2 overexpression (see Supplementary Datasets S2 and S3). In accordance with the paradigm: ‘Genes whose expression is anti-correlated with those studied might include members of a pathway whose action is opposed’,45 Gsk3β and Ankrd2 are both substrates of Akt2, but have opposite effects on myogenic differentiation. Under stress conditions triggering the production of ROS, Akt2 phosphorylation of Ankrd2 at Ser-99 causes its translocation to the nucleus where it negatively regulates muscle differentiation.8 On the other hand, Akt2 phosphorylation of Gsk3β causes its inactivation, thereby removing its inhibitory effect on differentiation and hypertrophy.46 We found that the inhibitory effect of the Ankrd2/p50 complex on Gsk3β is dependent on Akt2-mediated phosphorylation of Ankrd2 induced by hydrogen peroxide.
Based on our results, we propose a novel model in which activation of Akt2 in response to oxidative stress counteracts the toxic effects of ROS through inhibition of NF-κB activity via the repressor complex Ankrd2/p50 (Figure 8). Thus, when there is risk for tissue damage and resolution of inflammation needs to be assured, Ankrd2 may represent an important anti-inflammatory agent. This is particularly interesting as Ankrd2 expression has been shown to be positively correlated with physical exercise,3, 4, 47, 48 which induces an anti-inflammatory environment, counteracting the development of various chronic diseases,49 including dementia, diabetes, cardiovascular disease, stroke, and cancer. Thus, our results suggest a role of Ankrd2 in the protective effect of exercise through its ability to repress inflammatory responses.

Proposed model of a novel antioxidant defense system
Constitutively expressed inflammatory cytokines participate in the regulation of many important aspects of skeletal muscle function, including regeneration, hypertrophy, and whole body metabolism.11, 50 For example, although a chronic increase in the IL6 protein level is associated with pathological conditions such as atrophy,51 contracting muscle also produces and releases IL-6 to regulate glucose homeostasis.52 Our in vivo data suggest a further role of Ankrd2 in modulating the basal expression of cytokines under physiological conditions. In soleus muscle from Ankrd2 KO mice, calcineurin-stimulated transactivation of cytokines was found to be affected in a fiber type-specific manner as indicated by the downregulation of the key calcineurin-responsive genes Il6, Glut4, and Rcan1. This is particularly interesting as Ankrd2 mRNA is preferentially expressed in adult slow-twitch muscle fibers,34 and although we found no significant differences in fiber-type distribution between Ankrd2 KO and WT mice,23 our data reveal that slow muscle fibers of Ankrd2 KO mice are transcriptionally adapted to fiber-type switching toward a faster phenotype. This may appear contradictory, but in fact an increasing number of studies have shown mismatches in the relative proportion of mRNA and protein expression levels of myosin isoforms detected in slow–fast transitional fibers.53, 54, 55 The absent effect of Ankrd2 knockout on Akt, Gsk3β, and NF-κB activities in vivo may be because of compensatory mechanisms as the mouse model is a constitutive knockout.
This report provides a molecular mechanism for signal integration between Ankrd2 and NF-κB pathways, contributing to the understanding of the role of Ankrd2 as signaling molecule in skeletal muscle. The novel anti-inflammatory role of Ankrd2 through suppression of NF-κB transcription suggests its potential use as a target for the development of specific therapeutic strategies for combating inflammation in muscle.
Materials and Methods
Plasmid constructs
WT and Ser99Ala mutant FLAG-tagged human Ankrd2 expression vectors have been described previously.8 Full-length mouse p50 was amplified by PCR from mouse skeletal muscle cDNA using the primers: forward-EcoRI (5′-CGGAATTCCGGCATGGCAGACGATGATCCCTAC-3′) and reverse-SacII (5′-ATCGCCGCGGCGCTAATGGGTGACCCCTGCGTT-3′), digested with EcoRI/SacII, and inserted into a N-terminal FLAG-tag mammalian expression vector. The 3 × NF-κBluc reporter vector, containing a triplicate NF-κB response element, was generated by subcloning from a 3 × NF-κB-βgal reporter vector (kindly supplied by Professor Stefano Schiaffino, University of Padova, Padova, Italy) into the pGL3basic reporter vector (Promega, Madison, WI, USA). Briefly, the 230 bp DNA sequence containing the triplicate NF-κB response element was KpnI/HindIII excised from the 3 × NF-κBβgal plasmid and cloned into the same restriction sites of the pGL3basic reporter vector. Two luciferase reporter constructs were produced: shortPROwtluc and longPROwtluc containing 317 and 663 nucleotides upstream of the Ankrd2 ATG start codon, respectively. The reverse primer PROHindIII (5′-GGGAAGCTTCTGTTTCTGCAAGCCACAGGGC-3′) combined with shortPROXhoI (5′-CCGCTCGAGAGCCAGTTCCCAGCACTGAGGACAC-3′) and longPROXhoI (5′-CCGCTCGAGCCTGAGCTCTTAAAACAACACTAG-3′) were used for PCR amplification with Ankrd2 mouse genomic DNA as template. The PCR products were HindIII/XhoI digested and cloned into the pGL3-basic vector (Promega). Site-directed mutagenesis to remove the MyoD or the NF-κB-binding sequence was performed to generate three mutant reporters: shortPRO-MyoDluc, shortPRO-NF-κBluc, and longPRO-NF-κBluc. Oligonucleotide primers containing the desired mutations (MyoD-deleted primer: 5′-TGTCCGAGGTGAAGGAGTTGAGCTAGTAAGCTC-3′ and NF-κB-deleted primer: 5′- TGGCTTCCCATGCTCCCTGAGGTGAAGGTGACAG-3′) were used to amplify a mutation-containing replica of the WT plasmid. The PCR products were treated with DpnI before transformation into the E. coli DH10B strain. For generation of the luciferase reporter construct GSK3βPROluc, containing 765 nucleotides upstream of the ATG of the Gsk3β gene driving the firefly luciferase gene, PCR was performed on mouse genomic DNA using the primers: forward: 5′-CCGCTCGAGCGGTTGCCTGGTTCCCATCATG-3′ and reverse: 5′-GGGAAGCTTGATGGCTCTTCTTGAATC-3′. The PCR product was HindIII/XhoI digested and cloned into the pGL3-basic vector (Promega). All plasmids were confirmed by sequencing.
Generation of Ankrd2 KO mice
Genomic DNA was isolated from a mouse 129-SVJ genomic DNA library (Stratagene, La Jolla, CA, USA), using full-length Ankrd2 cDNA as a probe. The first 2 exons and half of exon 3 were replaced by cDNA encoding lacZ and a pGK neo cassette, thereby bringing the β-galactosidase cDNA under the control of the endogenous promoter while ablating the endogenous Ankrd2 gene (Supplementary Information, Supplementary Figure S1a). The targeting construct was verified by sequencing and linearized before electroporation into 129-SVJ-derived ES cells at the Transgenic Core Facility at the University of California, San Diego (La Jolla, CA, USA). ES clones were screened for homologous recombination by EcoRI digestion of ES cell DNA and Southern blot analysis (Supplementary Information, Supplementary Figure S1b) with the probe shown in Supplementary Information, Supplementary Figure S1a. Cells from two independent targeted ES cell clones were microinjected into C57BL/B6 blastocysts and transferred into pseudopregnant mice. Male chimeras resulting from the microinjections were mated to Black Swiss mice and gave rise to germline transmitted heterozygous mice that were subsequently interbred to generate homozygous mice. Offspring from intercrosses were genotyped by PCR analysis using mouse tail DNA and WT and KO allele-specific primers. The following primers were used: WT: sense: 5′-AACTTCGAAGATCCGCTCCTGG-3′; reverse: 5′-CATCAATGATCTCACGTCGCAG-3′ and KO: sense: 5′-CACACTGGACAGGCCTCTTTCC-3′; reverse: 5′-AGATGAAACGCCGAGTTAACGC-3′, giving rise to a 228-bp band from the WT allele and a 770-bp band from the KO allele. Successful ablation of the Ankrd2 gene was confirmed by northern (Supplementary Information, Supplementary Figure S1c) and western blot analyses (Supplementary Information, Supplementary Figure S1d) using polyclonal antibodies against Ankrd2.1 All animal procedures were in full compliance with the guidelines approved by the University of California San Diego Animal Care and Use Committee and the Italian Ministry of Health.
Adenovirus production
The adenovirus expressing Ankrd2-HA was created using the AdEasy strategy (Agilent Technologies, Santa Clara, CA, USA). The HA-tagged mouse Ankrd2 cDNA was cloned into the pAdTrack cytomegalovirus vector (Agilent Technologies). Subsequent steps were performed according to the manufacturer’s instructions. The adenoviral vectors contain two distinct promoters that independently drive the expression of the gene of interest and GFP. The mock plasmid expresses only GFP.
Cell cultures, DNA transfection, and adenoviral infection
The C2C12 mouse cell line was cultured as previously described.7 For infection, C2C12 cells were incubated with adenovirus at a multiplicity of infection (MOI) of 250 in growth or differentiation medium. The human embryonic kidney cell line (HEK293A) was cultured in DMEM supplemented with 10% heat-inactivated FBS. Cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Primary myoblasts were isolated from the limbs of 3-day-old neonatal WT and Ankrd2 KO mice using a protocol modified from Brodie and Sampson.56 Briefly, skinned mouse limbs were collected from neonatal pups, minced, and subjected to serial trypsinization (successive 25 min periods until all tissue was digested) in 0.05% trypsin solution B (Biological Industries, Kibbutz Beit-Haemek, Israel) in PBS by continuous stirring at 37°C. Cells were collected by centrifugation at 1000 × g for 5 min and resuspended in DMEM containing 20% FBS. After 30 min of preplating to remove fibroblasts, the supernatant was filtered through a nylon mesh and 1.0 × 106 cells/well were plated on collagen-coated six-well plates. Cells were proliferated in DMEM–high glucose containing 20% FBS and at a confluency of 80–95%, cell differentiation was induced by reducing serum levels to 2% horse serum. For adenoviral infection, cells were incubated with AdGFP or AdAnkrd2 at an MOI of 500 for 4 h in serum-free medium and collected 20 h after the initiation of infection. Adenovirus treatment was performed at three different stages: (1) proliferating myocytes at ∼50% confluency (proliferation stage); (2) proliferating myocytes at ∼90% confluency before induction of differentiation (fusion stage); and (3) differentiating cells 3 days after the induction of differentiation (differentiated stage).
RNA extraction and gene expression analyses
All microarray data are MIAME (Minimum Information About a Microarray Experiment) compliant. Raw data, normalized data, and detailed protocols are available in the GEO database (accession number GSE43500). Total RNA was isolated using TRIzol reagent (Invitrogen) following the manufacturer’s protocol. A Nanodrop ND-1000 (Agilent Technologies) was used for RNA quantification and the RNA 6000 Nano LabChip kit (Agilent Technologies) was used for RNA quality control in conjunction with an Agilent Bioanalyzer 2001. Only samples with an RNA Integrity Number (RIN) >9 were included in the study. For sample labeling, 50 ng of RNA was amplified and Cy3-labeled using the Low Input Quick Amp WT Labeling Kit, one-color (Agilent Technologies), in the presence of RNA Spike-In controls, following the manufacturer’s instructions. Briefly, RNA was mixed with 1 μl WT primer and 2 μl Agilent One-Color RNA Spike-In (1 : 20 000 dilution, Agilent Technologies) and denatured for 10 min at 65°C. First-Strand Buffer, DTT, dNTP mix, and AffinityScript RNaseBlock mix were added and cDNA synthesis was carried out for 2 h at 40°C. After 15 min at 70 °C to inactivate the enzymes, complementary RNA (cRNA) was generated using T7 RNA polymerase that amplifies target material and incorporates Cy3-labeled CTP. Run-off in vitro transcription reactions proceeded for 2 h at 40°C and the linearly amplified cRNA was purified with RNeasy mini columns (Qiagen, Hilden, Germany). For hybridization, fragmentation of 600 ng of Cy3-labeled cRNA was performed using the Agilent Hybridization Kit (Agilent Technologies) following the manufacturer’s instructions. Then, 40 μl of hybridization mix, composed of equal volumes of fragmented cRNA and 2 × GEx Hybridization Buffer HI-RPM, was dispensed onto one of the eight subarrays of the Agilent SurePrint G3 Mouse GE 8x60K Microarray (Agilent Technologies). Incubation proceeded for 17 h at 65°C at 10 r.p.m. in a Microarray Hybridization Oven (Agilent Technologies). Finally, slides were washed for 1 min at 22°C in Wash Solution 1 and subsequently for 1 min in Wash Solution 2, prewarmed to 37°C (Agilent Technologies). Microarrays were scanned using a GenPix 4000B Scanner (Agilent Technologies) for fluorescence detection at 3 μm resolution. Data were extracted from the scanned images using the Feature Extraction Software (Agilent Technologies) that also performed intra-array data normalization. The Expander software was applied to quantile normalize the complete series of experiments.57 Mean values were calculated for multiple probes with the same Agilent Probe ID. Differentially expressed genes were identified using the Significance Analysis of Microarray (SAM) test58 in the MultiExperiment Viewer (MeV, v4.8), a part of the TM4 Microarray Software Suite (Dana-Farber Cancer Institute, Boston, MA, USA).59 Two different false discovery rates (FDRs) were used: 0% (higher stringency) or 5% (lower stringency). KEGG pathway enrichment analysis was performed using the DAVID web server (http://david.abcc.ncifcrf.gov/home.jsp).60
Web-based-software
Enrichment of transcription factors in the promoter of DE genes was accomplished by using the oPOSSUM tool (http://opossum.cisreg.ca). To identify the occurrence and frequency of transcription factor binding sequences in the Gsk3β promoter, Genomatix software (http://www.genomatix.de/matinspector.html) and Patch tool (http://www.gene-regulation.com/cgi-bin/pub/programs/patch/bin/patch.cgi) were used.
Co-immunoprecipitation
Protein extracts from C2C12 cells were transfected with FLAG-tagged WT or Ser99Ala mutant human Ankrd2 or FLAG-tagged mouse p50 and incubated overnight at 4°C with anti-FLAG-tag antibody and resin provided by the IP/Co-IP Kit (Pierce, Rockford, IL, USA). Subsequently, immunoprecipitates were subjected to western blot analysis using antibodies against p50 (sc-114, Santa Cruz, Santa Cruz, CA, USA), p65 (sc-109, Santa Cruz), Ankrd2,26 and FLAG-tag.
Immunofluorescence microscopy
Proliferating (myoblasts) and differentiating (myotubes) C2C12 cells were grown on collagen-coated coverslips, fixed with 4% paraformaldehyde for 10 min, and subsequently processed for co-immunofluorescence staining using rabbit anti-p50 polyclonal antibody (sc-114, Santa Cruz) and a mouse monoclonal antibody specific for the Ankrd2 N-terminus26 as previously described. The immunostained samples were observed with a Leica (Solms, Germany) 5000 DFC300FX microscope at × 40 and × 100 magnifications.
Luciferase reporter assay
Luciferase reporter constructs were cotransfected with a modified pGL4.74 control reporter vector (Promega) in mouse C2C12 cells. The pGL4.74 vector was mutated by deletion of the thymidine kinase promoter to provide basal levels of Renilla luciferase expression in cotransfected cells. At 6 h after transfection, the medium was replaced with growth or differentiation-promoting medium and at 24, 48, and 144 h after transfection, cell lysates were prepared from proliferating and differentiating myoblasts. Luciferase and Renilla activity were measured on a TD-20/20 Single-Tube Luminometer (Turner BioSystems, Sunnyvale, CA, USA) using the Dual-Luciferase Reporter Assay System (Promega). Luciferase activity was normalized to Renilla activity to account for variations in transfection efficiency. Where indicated, transfected cells were treated with 250 μM H2O2 for 2, 4, and 6 h before collection. In some experiments, 2 μg/ml of LPS (Sigma-Aldrich, St. Louis, MO, USA) was added to the cells 24 or 48 h after transfection, whereafter cell lysates were prepared 24 h later.
Chromatin immunoprecipitation assays
ChIP assays were carried out using the ChIP assay kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. C2C12 myoblasts and myotubes at the fourth day of differentiation were fixed by adding formaldehyde solution into the culture medium to a final concentration of 1%, rinsed twice with ice-cold PBS, and lysed according to the manufacturer’s specifications. Sonication was performed on ice using a Sonic Dismembrator Model 300 (Thermo Fisher Scientific, Waltham, MA, USA) at a power of 50% for 3 cycles of sonication, each cycle for 30 s, followed by a 10-s break on ice. Chromatin complexes were immunoprecipitated with anti-Ankrd2, anti-p50, or anti-p65 antibodies and used as template for qRT-PCR. The primer sequences used are listed in Supplementary Information, Supplementary Table S2.
Real-time PCR
qRT-PCR based on SYBR Green chemistry (Finnzymes, Vantaa, Finland) was carried out as described previously.7 The primer sequences used are listed in Supplementary Information, Supplementary Table S3.
Protein extraction and western blot analysis
Total protein lysate from C2C12 cells was extracted by solubilization in RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP40, and 0.1% SDS) and protease inhibitor cocktail (Sigma-Aldrich), whereas mouse muscle was homogenized in the following lysis buffer: 50 mM Tris, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 0.5 mM DTT, 1% Triton X-100, 2% SDS, protease inhibitor cocktail (Roche, Indianapolis, IN, USA), and phosphatase inhibitor cocktail (Pierce). Western blot analyses were performed using antibodies against Ankrd2,26 phospho-Ankrd28 (Cenni V et al., manuscript in preperation), Gsk3β, phospho-Gsk3β (Ser-9), Akt, phospho-Akt (Ser-473), p65 and phospho-p65 (Ser-536), β-tubulin (all from Cell Signaling, Danvers, MA, USA), and β-actin (Sigma-Aldrich).
Accession codes
Abbreviations
- MARP:
-
muscle ankyrin repeat protein
- ROS:
-
reactive oxygen species
- KO:
-
knockout
- WT:
-
wild type
- qRT-PCR:
-
quantitative real-time PCR
- MB:
-
myoblast
- MT:
-
myotube
References
Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K et al. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol 2003; 333: 951–964.
Tsukamoto Y, Senda T, Nakano T, Nakada C, Hida T, Ishiguro N et al. Arpp, a new homolog of carp, is preferentially expressed in type 1 skeletal muscle fibers and is markedly induced by denervation. Lab Invest 2002; 82: 645–655.
Barash IA, Mathew L, Ryan AF, Chen J, Lieber RL . Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. Am J Physiol Cell Physiol 2004; 286: C355–C364.
McKoy G, Hou Y, Yang SY, Avelaira DV, Degens H, Goldspink G et al. Expression of Ankrd2 in fast and slow muscles and its response to stretch are consistent with a role in slow muscle function. J Appl Physiol 2005; 98: 2337–2343.
Hentzen ER, Lahey M, Peters D, Mathew L, Barash IA, Friden J et al. Stress-dependent and -independent expression of the myogenic regulatory factors and the MARP genes after eccentric contractions in rats. J Physiol 2006; 570 (Pt 1): 157–167.
Lehti TM, Silvennoinen M, Kivela R, Kainulainen H, Komulainen J . Effects of streptozotocin-induced diabetes and physical training on gene expression of titin-based stretch-sensing complexes in mouse striated muscle. Am J Physiol Endocrinol Metab 2007; 292: E533–E542.
Bean C, Facchinello N, Faulkner G, Lanfranchi G . The effects of Ankrd2 alteration indicate its involvement in cell cycle regulation during muscle differentiation. Biochim Biophys Acta 2008; 1783: 1023–1035.
Cenni V, Bavelloni A, Beretti F, Tagliavini F, Manzoli L, Lattanzi G et al. Ankrd2/ARPP is a novel Akt2 specific substrate and regulates myogenic differentiation upon cellular exposure to H(2)O(2). Mol Biol Cell 2011; 22: 2946–2956.
Kojic S, Medeot E, Guccione E, Krmac H, Zara I, Martinelli V et al. The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J Mol Biol 2004; 339: 313–325.
Pahl HL . Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999; 18: 6853–6866.
Pedersen BK, Febbraio MA . Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev 2008; 88: 1379–1406.
Hoffmann A, Levchenko A, Scott ML, Baltimore D . The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 2002; 298: 1241–1245.
Elsharkawy AM, Oakley F, Lin F, Packham G, Mann DA, Mann J . The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol 2010; 53: 519–527.
Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K et al. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell 1993; 72: 729–739.
Cao S, Zhang X, Edwards JP, Mosser DM . NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem 2006; 281: 26041–26050.
Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT et al. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 2006; 24: 563–574.
Gong R, Rifai A, Ge Y, Chen S, Dworkin LD . Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J Biol Chem 2008; 283: 7401–7410.
Steinbrecher KA, Wilson 3rd W, Cogswell PC, Baldwin AS . Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol 2005; 25: 8444–8455.
Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA, Sowers JR . Skeletal muscle insulin resistance: role of inflammatory cytokines and reactive oxygen species. Am J Physiol Regul Integr Comp Physiol 2008; 294: R673–R680.
Li YP, Schwartz RJ, Waddell ID, Holloway BR, Reid MB . Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J 1998; 12: 871–880.
De Rossi M, Bernasconi P, Baggi F, de Waal Malefyt R, Mantegazza R . Cytokines and chemokines are both expressed by human myoblasts: possible relevance for the immune pathogenesis of muscle inflammation. Int Immunol 2000; 12: 1329–1335.
Nagaraju K, Raben N, Merritt G, Loeffler L, Kirk K, Plotz P . A variety of cytokines and immunologically relevant surface molecules are expressed by normal human skeletal muscle cells under proinflammatory stimuli. Clin Exp Immunol 1998; 113: 407–414.
Barash IA, Bang ML, Mathew L, Greaser ML, Chen J, Lieber RL . Structural and regulatory roles of muscle ankyrin repeat protein family in skeletal muscle. Am J Physiol Cell Physiol 2007; 293: C218–C227.
Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS . Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev 2008; 22: 1490–1500.
Bai D, Ueno L, Vogt PK . Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 2009; 125: 2863–2870.
Pallavicini A, Kojic S, Bean C, Vainzof M, Salamon M, Ievolella C et al. Characterization of human skeletal muscle Ankrd2. Biochem Biophys Res Commun 2001; 285: 378–386.
Zhong S, Salomoni P, Pandolfi PP . The transcriptional role of PML and the nuclear body. Nat Cell Biol 2000; 2: E85–E90.
Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB . Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem 2004; 279: 39541–39554.
Mohamed JS, Lopez MA, Cox GA, Boriek AM . Anisotropic regulation of Ankrd2 gene expression in skeletal muscle by mechanical stretch. FASEB J 2010; 24: 3330–3340.
Bonizzi G, Karin M . The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25: 280–288.
Karin M, Ben-Neriah Y . Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 2000; 18: 621–663.
Bean C, Salamon M, Raffaello A, Campanaro S, Pallavicini A, Lanfranchi G . The Ankrd2, Cdkn1c and calcyclin genes are under the control of MyoD during myogenic differentiation. J Mol Biol 2005; 349: 349–366.
Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr. . NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 2000; 289: 2363–2366.
Chemello F, Bean C, Cancellara P, Laveder P, Reggiani C, Lanfranchi G . Microgenomic analysis in skeletal muscle: expression signatures of individual fast and slow myofibers. PLoS One 2011; 6: e16807.
Shi H, Scheffler JM, Zeng C, Pleitner JM, Hannon KM, Grant AL et al. Mitogen-activated protein kinase signaling is necessary for the maintenance of skeletal muscle mass. Am J Physiol Cell Physiol 2009; 296: C1040–C1048.
Banzet S, Koulmann N, Simler N, Birot O, Sanchez H, Chapot R et al. Fibre-type specificity of interleukin-6 gene transcription during muscle contraction in rat: association with calcineurin activity. J Physiol 2005; 566 (Pt 3): 839–847.
Smith JA, Collins M, Grobler LA, Magee CJ, Ojuka EO . Exercise and CaMK activation both increase the binding of MEF2A to the Glut4 promoter in skeletal muscle in vivo. Am J Physiol Endocrinol Metab 2007; 292: E413–E420.
Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN et al. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res 2000; 87: E61–E68.
Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M et al. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol 2008; 180: 787–802.
Laure L, Daniele N, Suel L, Marchand S, Aubert S, Bourg N et al. A new pathway encompassing calpain 3 and its newly identified substrate cardiac ankyrin repeat protein is involved in the regulation of the nuclear factor-kappaB pathway in skeletal muscle. FEBS J 2010; 277: 4322–4337.
Wu WS, Xu ZX, Hittelman WN, Salomoni P, Pandolfi PP, Chang KS . Promyelocytic leukemia protein sensitizes tumor necrosis factor alpha-induced apoptosis by inhibiting the NF-kappaB survival pathway. J Biol Chem 2003; 278: 12294–12304.
Remels AH, Langen RC, Gosker HR, Russell AP, Spaapen F, Voncken JW et al. PPARgamma inhibits NF-kappaB-dependent transcriptional activation in skeletal muscle. Am J Physiol Endocrinol Metab 2009; 297: E174–E183.
Jope RS, Yuskaitis CJ, Beurel E . Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res 2007; 32: 577–595.
Martin M, Rehani K, Jope RS, Michalek SM . Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 2005; 6: 777–784.
Qian J, Dolled-Filhart M, Lin J, Yu H, Gerstein M . Beyond synexpression relationships: local clustering of time-shifted and inverted gene expression profiles identifies new, biologically relevant interactions. J Mol Biol 2001; 314: 1053–1066.
Rochat A, Fernandez A, Vandromme M, Moles JP, Bouschet T, Carnac G et al. Insulin and wnt1 pathways cooperate to induce reserve cell activation in differentiation and myotube hypertrophy. Mol Biol Cell 2004; 15: 4544–4555.
Lehti M, Kivela R, Komi P, Komulainen J, Kainulainen H, Kyrolainen H . Effects of fatiguing jumping exercise on mRNA expression of titin-complex proteins and calpains. J Appl Physiol 2009; 106: 1419–1424.
Roberts MD, Childs TE, Brown JD, Davis JW, Booth FW . Early depression of Ankrd2 and Csrp3 mRNAs in the polyribosomal and whole tissue fractions in skeletal muscle with decreased voluntary running. J Appl Physiol 2012; 112: 1291–1299.
Pedersen BK . Exercise-induced myokines and their role in chronic diseases. Brain Behav Immun 2011; 25: 811–816.
Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P . Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab 2008; 7: 33–44.
Haddad F, Zaldivar F, Cooper DM, Adams GR . IL-6-induced skeletal muscle atrophy. J Appl Physiol 2005; 98: 911–917.
Febbraio MA, Pedersen BK . Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. FASEB J 2002; 16: 1335–1347.
Andersen JL, Schiaffino S . Mismatch between myosin heavy chain mRNA and protein distribution in human skeletal muscle fibers. Am J Physiol 1997; 272 (6 Pt 1): C1881–C1889.
Peuker H, Pette D . Quantitative analyses of myosin heavy-chain mRNA and protein isoforms in single fibers reveal a pronounced fiber heterogeneity in normal rabbit muscles. Eur J Biochem 1997; 247: 30–36.
Stevens L, Sultan KR, Peuker H, Gohlsch B, Mounier Y, Pette D . Time-dependent changes in myosin heavy chain mRNA and protein isoforms in unloaded soleus muscle of rat. Am J Physiol 1999; 277 (6 Pt 1): C1044–C1049.
Brodie C, Sampson SR . Nerve growth factor and fibroblast growth factor influence post-fusion expression of Na-channels in cultured rat skeletal muscle. J Cell Physiol 1990; 144: 492–497.
Sharan R, Maron-Katz A, Shamir R . CLICK and EXPANDER: a system for clustering and visualizing gene expression data. Bioinformatics 2003; 19: 1787–1799.
Tusher VG, Tibshirani R, Chu G . Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 2001; 98: 5116–5121.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 2003; 34: 374–378.
Huang da W, Sherman BT, Lempicki RA . Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009: 371–13.
Acknowledgements
We are grateful to Professor Oriano Marin, University of Padova, for the production of the phospho-Ankrd2 specific antibody. Dr. Chiara Romualdi is kindly acknowledged for assistance with statistical data analysis, Dr. Anna Raffaello for help with adenovirus vector production, and Alessandra Rodanò for the maintenance and genotyping of mice. This work was supported by grants from the Italian Ministry of University and Research (PRIN 2008YCBRJN and FIRB RBRN07BMCT) and the Marie Curie Actions FP7 for the Initial Training Network MUZIC (Muscle Z-disk Protein Complexes: from atomic structure to physiological function) to GL; from the Telethon Foundation, Italy (S07006TELA) to MLB and from the National Institute of Arthritis and Musculoskeletal and Skin to JC.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Bean, C., Verma, N., Yamamoto, D. et al. Ankrd2 is a modulator of NF-κB-mediated inflammatory responses during muscle differentiation. Cell Death Dis 5, e1002 (2014). https://doi.org/10.1038/cddis.2013.525
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.525
Keywords
This article is cited by
-
The titin N2B and N2A regions: biomechanical and metabolic signaling hubs in cross-striated muscles
Biophysical Reviews (2021)
-
Epigenetic regulation of VENTXP1 suppresses tumor proliferation via miR-205-5p/ANKRD2/NF-kB signaling in head and neck squamous cell carcinoma
Cell Death & Disease (2020)
-
Transcriptional reprogramming and phenotypic switching associated with the adaptation of Lactobacillus plantarum C2 to plant niches
Scientific Reports (2016)
-
Differential expression and localization of Ankrd2 isoforms in human skeletal and cardiac muscles
Histochemistry and Cell Biology (2016)
